Pivotal study showed durable responses, with a 56% overall response rate, a 43% complete response (remission) rate and a median duration of response of 1.5 years (18.4 months)
Given over a fixed period of time, Columvi provides patients with a treatment end date and potential time off treatment
Columvi is part of Roche's industry-leading portfolio of T-cell engaging bispecific antibodies in non-Hodgkin lymphoma, which also includes the recently approved Lunsumio to treat follicular lymphoma
Basel, 16 June 2023 – Roche (SIX: RO, ROG; OTCQX: RHHBY) announced today that the U.S. Food and Drug Administration (FDA) has approved Columvi® (glofitamab-gxbm) for the treatment of adult patients with relapsed or refractory (R/R) diffuse large B-cell lymphoma (DLBCL) not otherwise specified or large B-cell lymphoma (LBCL) arising from follicular lymphoma, after two or more lines of systemic therapy. This indication is approved under accelerated approval based on response rate and durability of response in the phase I/II NP30179 study. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial. Columvi will be available in the US in the coming weeks.
“People with diffuse large B-cell lymphoma who have gone through multiple lines of therapy have a poor prognosis and desperately need additional treatment options,” said Levi Garraway, M.D., Ph.D., Roche’s Chief Medical Officer and Head of Global Product Development. “As an off-the-shelf, fixed-duration treatment providing durable response rates, we believe Columvi could change the way this aggressive lymphoma is treated, reinforcing our dedication to bringing innovative treatment options to people with critical unmet needs.”
DLBCL is an aggressive, hard-to-treat disease and is the most common form of non-Hodgkin lymphoma in the US.2 While many people with DLBCL are responsive to treatment, the majority of those who relapse or are refractory to subsequent treatments have poor outcomes.3,4
“Patients with relapsed or refractory diffuse large B-cell lymphoma may experience rapid progression of their cancer and often urgently need an effective treatment option that can be administered without delay,” said Krish Patel, M.D., Director of the Lymphoma Program at the Swedish Cancer Institute in Seattle and investigator of the Columvi phase I/II NP30179 study. “Experience from clinical trials demonstrates that Columvi can provide patients with relapsed or refractory diffuse large B-cell lymphoma a chance for complete remission with a fixed duration immunotherapy and that such remissions can potentially be sustained after the end of their treatment.”
The FDA accelerated approval is based on positive results from the phase I/II NP30179 study of Columvi given as a fixed course for 8.5 months in 132 patients with DLBCL who had relapsed or were refractory to prior therapies, including about one-third (30%) who had received prior CAR T-cell therapy. Additionally, 83% were refractory to their most recent therapy. Results showed patients treated with fixed-duration Columvi achieved durable remission, with 56% of patients achieving an overall response (OR; 74/132 [95% confidence interval (CI): 47-65]) and 43% of patients achieving a complete response (CR; 57/132 [95% CI: 35-52]). Over two-thirds of those who responded continued to respond for at least nine months (68.5% [95% CI: 56.7-80.3]). The OR rate is the combination of CR rate (a disappearance of all signs and symptoms of cancer) and partial response rate (a decrease in the amount of cancer in the body). The median duration of response was 1.5 years (18.4 months [95% CI: 11.4-not estimable]).1 Data from the NP30179 study were recently published in the New England Journal of Medicine.
Among 145 patients who received Columvi in the study, the most common adverse events (AEs) were cytokine release syndrome (CRS; 70%), which may be serious or life-threatening, musculoskeletal pain (21%), fatigue (20%) and rash (20%). CRS was generally low grade (52% experienced Grade 1, and 14% experienced Grade 2). 1
Columvi is the first and only CD20xCD3 T-cell engaging bispecific antibody for the treatment of R/R DLBCL that is given for a defined period of time, unlike treat-to-progression approaches where treatment is given indefinitely until the cancer progresses or the therapy cannot be tolerated, whichever occurs first. Designed to be completed in approximately 8.5 months, Columvi offers people with R/R DLBCL a target end date for their course of treatment and the possibility of a treatment-free period. Additionally, Columvi is a chemotherapy-free treatment option that is off-the-shelf and ready for infusion.
Columvi is part of Roche’s broad and industry-leading CD20xCD3 T-cell-engaging bispecific antibody clinical development programme. This includes the phase III STARGLO study evaluating Columvi in combination with gemcitabine and oxaliplatin (GemOx) versus MabThera®/Rituxan® (rituximab) in combination with GemOx in patients with DLBCL who have been treated with one or more previous therapies and are ineligible for autologous stem cell transplant. Roche’s haematology bispecific antibody portfolio also includes Lunsumio® (mosunetuzumab), which was granted accelerated approval by the FDA in December 2022 for the treatment of adult patients with R/R follicular lymphoma (FL) after two or more lines of systemic therapy. Roche is exploring the potential of both Columvi and Lunsumio as monotherapies and in combination with other therapies, including Polivy® (polatuzumab vedotin), in earlier lines of treatment with the goal of providing patients with long-lasting outcomes. This robust development programme includes two phase III studies: CELESTIMO, investigating Lunsumio plus lenalidomide in second line plus (2L+) FL, and SUNMO, investigating Lunsumio plus Polivy in 2L+ DLBCL. Columvi received its first worldwide approval in Canada, and the European Medicines Agency’s Committee for Medicinal Products for Human Use recently granted a positive opinion recommending its approval.
Columvi is a CD20xCD3 T-cell-engaging bispecific antibody designed to target CD3 on the surface of T-cells and CD20 on the surface of B-cells. Columvi was designed with a novel 2:1 structural format. This T-cell-engaging bispecific antibody is engineered to have one region that binds to CD3, a protein on T-cells, a type of immune cell, and two regions that bind to CD20, a protein on B-cells, which can be healthy or malignant. This dual-targeting brings the T-cell in close proximity to the B-cell, activating the release of cancer cell-killing proteins from the T-cell. A clinical development programme for Columvi is ongoing, investigating the molecule as a monotherapy and in combination with other medicines for the treatment of people with B-cell non-Hodgkin lymphomas, including diffuse large B-cell lymphoma and other blood cancers.
The NP30179 study [NCT03075696] is a phase I/II, multicentre, open-label, dose-escalation and expansion study evaluating the safety, efficacy and pharmacokinetics of Columvi® (glofitamab-gxbm) in people with relapsed or refractory diffuse large B-cell lymphoma. Outcome measures include complete response rate by an independent review committee (primary endpoint), overall response rate, duration of response, progression-free survival, safety, and tolerability (secondary endpoints).
DLBCL is the most common form of non-Hodgkin lymphoma (NHL), accounting for about one in three cases of NHL.2 DLBCL is an aggressive (fast-growing) type of NHL.2 While it is generally responsive to treatment in the frontline, as many as 40% of people will relapse or have refractory disease, at which time salvage therapy options are limited and survival is short.3,4 Improving treatments earlier in the course of the disease and providing needed alternative options could help to improve long-term outcomes. Approximately 160,000 people worldwide are estimated to be diagnosed with DLBCL each year.
Roche has been developing medicines for people with malignant and non-malignant blood diseases for more than 20 years; our experience and knowledge in this therapeutic area runs deep. Today, we are investing more than ever in our effort to bring innovative treatment options to patients across a wide range of haematologic diseases. Our approved medicines include MabThera®/Rituxan® (rituximab), Gazyva®/Gazyvaro® (obinutuzumab), Polivy® (polatuzumab vedotin), Venclexta®/Venclyxto® (venetoclax) in collaboration with AbbVie, Hemlibra® (emicizumab), Lunsumio® (mosunetuzumab) and Columvi® (glofitamab). Our pipeline of investigational haematology medicines includes a T-cell-engaging bispecific antibody cevostamab, targeting both FcRH5 and CD3; Tecentriq® (atezolizumab), a monoclonal antibody designed to bind with PD-L1 and crovalimab, an anti-C5 antibody engineered to optimise complement inhibition. Our scientific expertise, combined with the breadth of our portfolio and pipeline, also provides a unique opportunity to develop combination regimens that aim to improve the lives of patients even further.
Roche is a global pioneer in pharmaceuticals and diagnostics focused on advancing science to improve people’s lives. The combined strengths of pharmaceuticals and diagnostics under one roof have made Roche the leader in personalised healthcare – a strategy that aims to fit the right treatment to each patient in the best way possible.
Roche is the world’s largest biotech company, with truly differentiated medicines in oncology, immunology, infectious diseases, ophthalmology and diseases of the central nervous system. Roche is also the world leader in in vitro diagnostics and tissue-based cancer diagnostics, and a frontrunner in diabetes management.
Founded in 1896, Roche continues to search for better ways to prevent, diagnose and treat diseases and make a sustainable contribution to society. The company also aims to improve patient access to medical innovations by working with all relevant stakeholders. More than thirty medicines developed by Roche are included in the World Health Organization Model Lists of Essential Medicines, among them life-saving antibiotics, antimalarials and cancer medicines. Moreover, for the twelfth consecutive year, Roche has been recognised as one of the most sustainable companies in the Pharmaceuticals Industry by the Dow Jones Sustainability Indices (DJSI).
The Roche Group, headquartered in Basel, Switzerland, is active in over 100 countries and in 2020 employed more than 100,000 people worldwide. In 2020, Roche invested CHF 12.2 billion in R&D and posted sales of CHF 58.3 billion. Genentech, in the United States, is a wholly owned member of the Roche Group. Roche is the majority shareholder in Chugai Pharmaceutical, Japan
Every year, over 600,000 women worldwide are diagnosed with cervical cancer and over 340,000 die from this preventable disease, caused by infection with human papillomavirus (HPV). Nine out of 10 women who die from cervical cancer live in low- and lower-middle income countries (LMICs).
WHO prequalification enables LMICs to use the cobas® HPV test in their national cervical cancer elimination programs, increasing access to the patients who need it most.
Establishing screening programs helps prevent and detect cervical cancer, which is especially important in areas with limited healthcare resources where patients are often diagnosed with the disease at late stages.
Basel, 13 June 2023 - Roche (SIX: RO, ROG; OTCQX: RHHBY) today announced that the cobas® HPV test for use on the cobas® 6800/8800 Systems has been awarded World Health Organization (WHO) prequalification. WHO prequalification expands the availability of this critical HPV screening tool in countries that rely on the global organisation’s list in making purchasing and implementation decisions.
Screening for Human Papillomavirus (HPV) can help identify women who are at risk of developing cervical cancer, so that the disease can be treated early, before invasive cancer has a chance to develop. In poorer countries, women are often diagnosed with cervical cancer at a more advanced stage, where the opportunity for a cure is low.
“The elimination of cervical cancer is within reach. Roche is committed to working with governments, non-profit organisations and funders to help build sustainable cervical cancer elimination programs so that women, no matter where in the world they live, no longer die from this preventable disease,” said Matt Sause, CEO of Roche Diagnostics. “Today’s action, combined with our recently-launched HPV-self sampling solution, further expands access to HPV screening in countries with limited healthcare resources.”
The WHO strategy for global elimination of cervical cancer lists the following three target goals to reach by 20302:
90% of girls should be fully vaccinated with HPV vaccine by 15 years of age;
70% of women should be screened using a high-performance test by age 35, and again by age 45;
90% of those identified with cervical disease should receive appropriate treatment.
The cobas® HPV test is already part of the Roche Global Access Program, which aims to improve access to cost-effective resources, implement scale-up programs, and contribute to the elimination of diseases in the regions with the greatest need. WHO prequalification helps expand that access and provides healthcare professionals with greater confidence that their clinical decisions will be based on accurate, reliable results.
In 2014, Roche first launched its Global Access Program to support the UNAIDS 2020 targets to address the HIV/AIDS epidemic. Since then, the program was expanded to include solutions for other high-burden diseases such as Tuberculosis, Hepatitis B and C, and cervical cancer. Most recently, in response to the COVID-19 pandemic, the SARS-CoV-2 test was included into the program.
The continual expansion of test offerings highlights Roche's commitment to eliminate cervical cancer and other high burden infectious diseases for patients living in resource-constrained settings with limited access.
Any laboratory that implements a Roche instrument system gains the ability to scale up testing across multiple disease areas, thus improving cost and resource efficiency. An integrated approach supports national programs focused on increasing access to diagnostic testing, to help manage or reduce the impact of preventable disease for patients.
The cobas® HPV test is indicated for use for routine cervical cancer screening as per professional medical guidelines, including HPV primary screening, co-testing (or adjunctive screen) with cytology, and for triage of women with abnormal cytology, to assess the risk for cervical precancer and cancer. The cobas® HPV test detects the high-risk HPV types 16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59, 66, and 68.
In June 2022, Roche further improved access for women when it launched an HPV self-sampling solution in countries accepting the CE mark. The solution enables a patient to privately and confidently collect her own sample following instruction from a healthcare worker. The clinically-validated vaginal sample is then analysed with the Roche cobas® HPV test on a Roche molecular instrument.
Cervical cancer screening using the cobas® HPV test is clinically validated in large, FDA registrational trials for use on cobas® Systems, and the assay individually identifies the presence of the DNA of HPV genotypes 16 and 18 – the two genotypes responsible for about 70 percent of all cervical cancers – and reporting the 12 other high-risk HPV types as a combined result, all in one test and from one patient sample. More information about the cobas® HPV tests is available at diagnostics.roche.com/cervicalcancer.
The fully automated cobas® 6800/8800 Systems offer the fastest time to results, providing up to 96 results in about three hours and 384 results for the cobas® 6800 System and 1,056 results for the cobas® 8800 System in an eight hour shift.
Roche is a global pioneer in pharmaceuticals and diagnostics focused on advancing science to improve people’s lives. The combined strengths of pharmaceuticals and diagnostics under one roof have made Roche the leader in personalised healthcare – a strategy that aims to fit the right treatment to each patient in the best way possible.
Roche is the world’s largest biotech company, with truly differentiated medicines in oncology, immunology, infectious diseases, ophthalmology and diseases of the central nervous system. Roche is also the world leader in in vitro diagnostics and tissue-based cancer diagnostics, and a frontrunner in diabetes management.
Founded in 1896, Roche continues to search for better ways to prevent, diagnose and treat diseases and make a sustainable contribution to society. The company also aims to improve patient access to medical innovations by working with all relevant stakeholders. More than thirty medicines developed by Roche are included in the World Health Organization Model Lists of Essential Medicines, among them life-saving antibiotics, antimalarials and cancer medicines. Moreover, for the twelfth consecutive year, Roche has been recognised as one of the most sustainable companies in the Pharmaceuticals Industry by the Dow Jones Sustainability Indices (DJSI).
The Roche Group, headquartered in Basel, Switzerland, is active in over 100 countries and in 2020 employed more than 100,000 people worldwide. In 2020, Roche invested CHF 12.2 billion in R&D and posted sales of CHF 58.3 billion. Genentech, in the United States, is a wholly owned member of the Roche Group. Roche is the majority shareholder in Chugai Pharmaceutical, Japan
The COMMODORE 2 study demonstrated that subcutaneous crovalimab every four weeks was non-inferior to intravenous eculizumab every two weeks, with comparable safety, in people new to C5 inhibitors
Monthly self-administration of subcutaneous crovalimab has the potential to address the high burden of a disease that requires lifelong treatment including in settings where access to current C5 inhibitors is limited
The COMMODORE 1 study in people switching from currently approved C5 inhibitors supported the consistent benefit-risk profile of crovalimab as seen in the COMMODORE 2 study
Basel, 09 June 2023 - Roche (SIX: RO, ROG; OTCQX: RHHBY) announced today that positive results from the global phase III COMMODORE 1 and 2 studies, evaluating the efficacy and safety of crovalimab, an investigational, novel anti-C5 recycling monoclonal antibody, compared to eculizumab, a current standard of care in paroxysmal nocturnal haemoglobinuria (PNH) were presented at the European Hematology Association (EHA) Hybrid Congress, taking place in Frankfurt, Germany on 8-11 June 2023.
“With the option for subcutaneous self-administration, crovalimab could help meet the lifelong needs of people living with PNH and their caregivers,” said Levi Garraway, M.D., Ph.D., Roche’s Chief Medical Officer and Head of Global Product Development. “Data from the COMMODORE studies will be submitted to regulatory authorities around the world.”
PNH is a rare and life-threatening blood condition, in which red blood cells are destroyed by the complement system — part of the innate immune system — causing symptoms such as anaemia, fatigue, blood clots and kidney disease.4 C5 inhibitors have been shown to be effective in treating the condition.5 Crovalimab has been engineered to be recycled within the bloodstream, enabling sustained complement inhibition through low dose, subcutaneous (SC) administration every four weeks.
In the COMMODORE 2 study, 79.3% (95% CI: 72.9, 84.5) of participants randomised to be treated with crovalimab achieved haemolysis control from week five to week 25 compared with 79.0% (95% CI: 69.7, 86.0) with eculizumab. Additionally, 65.7% (95% CI: 56.9, 73.5) achieved transfusion avoidance (TA) from baseline to week 25 with crovalimab and 68.1% (95% CI: 55.7, 78.5) with eculizumab. TA is defined as people who become transfusion-free and do not require transfusion per protocol-specified guidelines. Blood transfusion requirements are important clinical measures of haemolysis caused by complement dysregulation in PNH. A clinically meaningful improvement in FACIT-Fatigue score from baseline to week 25 occurred in both arms, with a numerically greater improvement with crovalimab (adjusted mean change 7.8 (95% Cl: 6.5, 9.1)), versus eculizumab (adjusted mean change 5.2 (95% Cl: 3.4, 6.9)).
Adverse events (AEs) occurred in 78% of participants treated with crovalimab and 80% treated with eculizumab in the COMMODORE 2 study. Serious infections occurred in 3% of participants treated with crovalimab and 7% of participants treated with eculizumab, with no meningococcal infections. The most common AE, occurring in 16% of people treated with crovalimab and 13% of people treated with eculizumab was an infusion-related reaction. One participant in each arm experienced an AE that led to treatment discontinuation.
The results from the COMMODORE 1 study indicate that crovalimab maintained disease control in people switching from currently approved complement inhibitors.3 The data support the consistent benefit-risk profile of crovalimab, as well as SC administration with the option to self-administer, as seen in the COMMODORE 2 study.
Roche also presented preliminary data from the COMMODORE Burden of Illness study, which suggest that despite currently available C5 inhibitor treatments, people with PNH continue to experience substantial burden of disease, which can translate into diminished quality of life and considerable costs. These data suggest that people with PNH may benefit from alternative treatment options.
Global phase III data from the COMMODORE 1 and 2 studies in PNH will be submitted to regulatory authorities around the world. Positive data from a third phase III study evaluating crovalimab in PNH, the COMMODORE 3 study in China, were presented at the American Society of Hematology (ASH) Annual Meeting and Exposition on 10 December 2022. Data from the COMMODORE 3 study have been submitted via China’s Centre for Drug Evaluation Breakthrough Therapy Designation pathway. This submission has been accepted under Priority Review for approval consideration by China’s National Medical Products Administration.
Crovalimab is an investigational, novel anti-C5 recycling monoclonal antibody designed to block the complement system – a vital part of the innate immune system that acts as the body’s first line of defence against infection. Crovalimab, which was created by Chugai Pharmaceutical Co., Ltd, has been engineered to address the medical needs of people living with complement-mediated diseases, including providing patients with a potential self-administration option.
Crovalimab works by binding to C5, blocking the last step of the complement cascade and is also recycled within the bloodstream, enabling rapid and sustained complement inhibition.6,7 Crovalimab’s recycling properties also enables low dose SC administration every four weeks. In addition, crovalimab binds to a different C5 binding site from current treatments, which has the potential to provide a treatment option for people with specific C5 gene mutations, who do not respond to current therapies.6 It is also being evaluated in atypical haemolytic uraemic syndrome, sickle cell disease, and other complement mediated diseases.
The COMMODORE 2 study is a phase III, randomised, open-label study evaluating the efficacy and safety of crovalimab versus eculizumab in people with paroxysmal nocturnal haemoglobinuria (PNH) who have not been treated previously with C5 inhibitors. The 204 adults enrolled in the study were randomised in a 2:1 ratio, to be treated with either subcutaneous (SC) crovalimab every four weeks or intravenous (IV) eculizumab every two weeks. The six participants who were less than 18 years old were included in a non-randomised arm, to be treated with SC crovalimab every four weeks.9
The COMMODORE 1 study is a phase III, randomised, open-label study evaluating the safety of crovalimab in people with PNH switching from currently approved C5 inhibitors. The study included 89 people (18 years of age or older) currently treated with eculizumab, randomised in a 1:1 ratio to be treated with either SC crovalimab every four weeks or IV eculizumab every two weeks. In a non-randomised arm, the study also included paediatrics (<18 years of age) currently treated with eculizumab, people currently treated with ravulizumab, people currently treated with off-label doses of eculizumab (higher than the approved dose for PNH: more than 900mg per dose and/or more frequently than every two weeks), or people with known mutations in the C5 gene who do not respond to current therapies.
Roche is a global pioneer in pharmaceuticals and diagnostics focused on advancing science to improve people’s lives. The combined strengths of pharmaceuticals and diagnostics under one roof have made Roche the leader in personalised healthcare – a strategy that aims to fit the right treatment to each patient in the best way possible.
Roche is the world’s largest biotech company, with truly differentiated medicines in oncology, immunology, infectious diseases, ophthalmology and diseases of the central nervous system. Roche is also the world leader in in vitro diagnostics and tissue-based cancer diagnostics, and a frontrunner in diabetes management.
Founded in 1896, Roche continues to search for better ways to prevent, diagnose and treat diseases and make a sustainable contribution to society. The company also aims to improve patient access to medical innovations by working with all relevant stakeholders. More than thirty medicines developed by Roche are included in the World Health Organization Model Lists of Essential Medicines, among them life-saving antibiotics, antimalarials and cancer medicines. Moreover, for the twelfth consecutive year, Roche has been recognised as one of the most sustainable companies in the Pharmaceuticals Industry by the Dow Jones Sustainability Indices (DJSI).
The Roche Group, headquartered in Basel, Switzerland, is active in over 100 countries and in 2020 employed more than 100,000 people worldwide. In 2020, Roche invested CHF 12.2 billion in R&D and posted sales of CHF 58.3 billion. Genentech, in the United States, is a wholly owned member of the Roche Group. Roche is the majority shareholder in Chugai Pharmaceutical, Japan
Fenebrutinib is an investigational, potent and highly selective oral Bruton’s tyrosine kinase (BTK) inhibitor, the only reversible BTK inhibitor currently in Phase III multiple sclerosis (MS) trials
Phase II study met its primary and secondary endpoints by reducing the total number of new gadolinium-enhancing T1 brain lesions and significantly reducing the total number of new or enlarging T2 brain lesions compared to placebo
The safety profile of fenebrutinib was consistent with previous and ongoing clinical trials across more than 2,400 people to date
Basel, 17 May 2023 - Roche (SIX: RO, ROG; OTCQX: RHHBY) announced today positive results from the Phase II FENopta study evaluating investigational oral fenebrutinib in adults with relapsing forms of multiple sclerosis (RMS). The study met its primary and secondary endpoints, showing oral fenebrutinib significantly reduced magnetic resonance imaging (MRI) markers of MS disease activity in the brain compared to placebo. Additionally, pre-clinical data have shown fenebrutinib to be potent and highly selective, and it is the only reversible inhibitor currently in Phase III trials for MS.
“I am encouraged by this clinical data for fenebrutinib, which is important news for people living with MS,” said Levi Garraway, M.D., Ph.D., Roche’s Chief Medical Officer and Head of Global Product Development. “Fenebrutinib’s mechanism of action which can inhibit both B cells and microglia, has the potential to both reduce MS disease activity, such as relapses, and also impact disease progression.’’
Fenebrutinib significantly reduced the total number of new gadolinium-enhancing T1 brain lesions compared to placebo, the primary endpoint of the trial (p=0.0022). Additionally, fenebrutinib significantly reduced the total number of new or enlarging T2 brain lesions compared to placebo, a secondary endpoint. Furthermore, a higher proportion of patients treated with fenebrutinib were free from any new gadolinium-enhancing T1 brain lesions and new or enlarging T2-weighted brain lesions compared to placebo. T1 lesions, as measured by MRI, are a marker of active inflammation and T2 lesions represent the amount of disease burden or lesion load.
The safety profile of fenebrutinib was consistent with previous and ongoing fenebrutinib clinical trials across more than 2,400 people to date. There were no new safety concerns identified in the FENopta study.
Detailed results will be shared at an upcoming medical meeting. The Phase III fenebrutinib clinical trial programme in RMS and primary progressive MS (PPMS) is ongoing.
Fenebrutinib is an investigational oral, reversible and non-covalent Bruton’s tyrosine kinase (BTK) inhibitor that blocks the function of BTK. BTK, also known as tyrosine-protein kinase BTK, is an enzyme that regulates B-cell development and activation and is also involved in the activation of innate immune system myeloid lineage cells, such as macrophages and microglia. Pre-clinical data have shown fenebrutinib to be potent and highly selective, and it is the only reversible inhibitor currently in Phase III trials for MS. Fenebrutinib has been shown to be 130 times more selective for BTK vs. other kinases. These design features may be important as the high selectivity and reversibility can potentially reduce off-target effects of a molecule and contribute to long-term safety outcomes.
Fenebrutinib is a dual inhibitor of both B-cell and microglia activation. This dual inhibition has the potential to reduce both MS disease activity and progression, thereby addressing the key unmet medical need in people living with MS. The Phase III programme includes two identical trials in RMS (FENhance 1 & 2) with an active teriflunomide comparator and one trial in primary progressive MS (PPMS) (FENtrepid) in which fenebrutinib is being evaluated against OCREVUS® (ocrelizumab). To date, more than 2,400 patients and healthy volunteers have been treated with fenebrutinib in Phase I, II and III clinical programmes across multiple diseases, including MS and other autoimmune disorders.
The FENopta study is a global Phase II, randomised, double-blind, placebo-controlled 12-week study to investigate the efficacy, safety and pharmacokinetics of fenebrutinib in 109 adults aged 18-55 years with RMS. The primary endpoint is the total number of new gadolinium-enhancing T1 lesions as measured by MRI scans of the brain at 4, 8 and 12 weeks. Secondary endpoints include the number of new or enlarging T2-weighted lesions as measured by MRI scans of the brain at 4, 8 and 12 weeks, and the proportion of patients free from any new gadolinium-enhancing T1 lesions and new or enlarging T2-weighted lesions as measured by MRI scans of the brain at 4, 8 and 12 weeks. The goal of the FENopta study is to characterise the effect of fenebrutinib on MRI and soluble biomarkers of disease activity and progression, and it includes an optional substudy to measure cerebrospinal fluid biomarkers of neuronal injury. Patients who complete the double-blind period are eligible for an open-label extension study.
Multiple sclerosis (MS) is a chronic disease that affects more than 2.8 million people worldwide. MS occurs when the immune system abnormally attacks the insulation and support around nerve cells (myelin sheath) in the central nervous system (brain, spinal cord and optic nerves), causing inflammation and consequent damage. This damage can cause a wide range of symptoms, including muscle weakness, fatigue and difficulty seeing, and may eventually lead to disability. Most people with MS experience their first symptom between 20 and 40 years of age, making the disease the leading cause of non-traumatic disability in younger adults.
People with all forms of MS experience disease progression – permanent loss of nerve cells in the central nervous system – from the beginning of their disease even if their clinical symptoms aren’t apparent or don’t appear to be getting worse. Delays in diagnosis and treatment can negatively impact people with MS, in terms of their physical and mental health, and contribute to the negative financial impact on the individual and society. An important goal of treating MS is to slow, stop and ideally prevent disease activity and progression as early as possible.
Relapsing-remitting MS (RRMS) is the most common form of the disease and is characterised by episodes of new or worsening signs or symptoms (relapses) followed by periods of recovery. Approximately 85% of people with MS are initially diagnosed with RRMS. The majority of people who are diagnosed with RRMS will eventually transition to secondary progressive MS (SPMS), in which they experience steadily worsening disability over time. Relapsing forms of MS (RMS) include people with RRMS and people with SPMS who continue to experience relapses. Primary progressive MS (PPMS) is a debilitating form of the disease marked by steadily worsening symptoms but typically without distinct relapses or periods of remission. Approximately 15% of people with MS are diagnosed with the primary progressive form of the disease. Until the FDA approval of OCREVUS, there had been no FDA-approved treatments for PPMS.
Neuroscience is a major focus of research and development at Roche. Our goal is to pursue ground-breaking science to develop new treatments that help improve the lives of people with chronic and potentially devastating diseases. Roche and Genentech are investigating more than a dozen medicines for neurological disorders, including MS, spinal muscular atrophy, neuromyelitis optica spectrum disorder, Alzheimer’s disease, Huntington’s disease, Parkinson’s disease, acute ischemic stroke, Duchenne muscular dystrophy and Angelman syndrome. Our MS franchise also includes OCREVUS, which has been approved in more than 100 countries globally with 300,000 people treated to date. Together with our partners, we are committed to pushing the boundaries of scientific understanding to solve some of the most difficult challenges in neuroscience today.
Roche is a global pioneer in pharmaceuticals and diagnostics focused on advancing science to improve people’s lives. The combined strengths of pharmaceuticals and diagnostics under one roof have made Roche the leader in personalised healthcare – a strategy that aims to fit the right treatment to each patient in the best way possible.
Roche is the world’s largest biotech company, with truly differentiated medicines in oncology, immunology, infectious diseases, ophthalmology and diseases of the central nervous system. Roche is also the world leader in in vitro diagnostics and tissue-based cancer diagnostics, and a frontrunner in diabetes management.
Founded in 1896, Roche continues to search for better ways to prevent, diagnose and treat diseases and make a sustainable contribution to society. The company also aims to improve patient access to medical innovations by working with all relevant stakeholders. More than thirty medicines developed by Roche are included in the World Health Organization Model Lists of Essential Medicines, among them life-saving antibiotics, antimalarials and cancer medicines. Moreover, for the twelfth consecutive year, Roche has been recognised as one of the most sustainable companies in the Pharmaceuticals Industry by the Dow Jones Sustainability Indices (DJSI).
The Roche Group, headquartered in Basel, Switzerland, is active in over 100 countries and in 2020 employed more than 100,000 people worldwide. In 2020, Roche invested CHF 12.2 billion in R&D and posted sales of CHF 58.3 billion. Genentech, in the United States, is a wholly owned member of the Roche Group. Roche is the majority shareholder in Chugai Pharmaceutical, Japan
Almost half of the world’s population has no or limited access to diagnostics
This situation is especially acute in low- and middle-income countries (LMICs) where diagnostics plays a critical role for treating and containing the spread of infectious diseases such as Tuberculosis (TB) and HIV
New partnership between Roche and the Global Fund supports low- and middle-income countries in broadening access to diagnostics, helping millions of previously undiagnosed people with TB and HIV get diagnosed and eventually treated
Basel, 12 May 2022 - Roche (SIX: RO, ROG; OTCQX: RHHBY), through its Global Access Program, and The Global Fund to Fight AIDS, Tuberculosis and Malaria are joining forces to build and strengthen diagnostic capacity and pandemic preparedness in low-and middle-income countries fighting against HIV and tuberculosis (TB).
About 2 billion people worldwide are estimated to be infected with tuberculosis, with 95% of TB deaths occurring in LMICs. Of more than 37 Million people living with HIV globally, over 6 Million people are undiagnosed. The COVID-19 pandemic has disrupted many HIV and TB programs, resulting in HIV testing rates falling by 22% and an estimated additional 100 000 deaths from tuberculosis in LMICs in 2020.1,2
Roche and the Global Fund have joined forces to improve the diagnosis of HIV and TB in LMICs by building local capacity to tackle fundamental infrastructure challenges for generating and delivering diagnostic results and managing healthcare waste.
This includes building effective processes to collect, transport, test samples and return the results to patients for timely clinical interventions, as well as addressing challenges arising from a lack of network infrastructure, workforce capacity, access to roads, and IT systems. The partnership will also include novel approaches to reduce the environmental and economic burden of healthcare waste generated during the testing process itself and the disposal of instruments and medical devices at the end of its/their useful life.
“Roche is excited to join forces with the Global Fund and their partners to support countries in developing critical diagnostic networks in the global fight against HIV and TB.”, said Thomas Schinecker, CEO of Roche Diagnostics." Connecting our experts with critical local stakeholders, we are aiming to help build sustainable solutions that could be scaled across countries."
“Getting people to test for HIV and TB is fundamental to containing transmission and enrolling people on treatment, which are crucial steps to saving lives and ending these diseases as public health threats,” said Peter Sands, Executive Director of the Global Fund. “We are pleased to partner with Roche in expanding access to diagnostics tools for HIV and TB. These efforts will strengthen the fight against these diseases and help the world prepare better for future pandemics.”
Through collaboration with the Global Fund, Ministries of Health and country-based partners, Roche will first support assessments and implementation of new technologies and knowledge transfer in 2 to 3 pilot countries, with the ambition to scale up and expand support in 10 countries over the next five years.
In 2014, Roche first launched its Global Access Program to support the UNAIDS 2020 targets to address the HIV/AIDS epidemic. Since then, the program was expanded to include solutions for other high-burden diseases such as Tuberculosis (TB), Hepatitis B and C (HBV and HCV), and Human Papillomavirus (HPV). Most recently, in response to the COVID-19 pandemic, the SARS-CoV-2 test was included into the program. The program is designed to support end-to-end, sustainable, local solutions that build capacity and strengthen healthcare systems, with a focus on diagnostics and laboratory networks.
The continual expansion of offerings highlights Roche's commitment to eliminate high burden infectious diseases for patients living in resource-constrained settings with limited access.
Any laboratory that implements a Roche instrument system gains the ability to scale up testing across multiple disease areas, thus increasing efficiency with respect to limited resources or time. An integrated approach supports national programs focused on increasing access to diagnostic testing, to help manage or reduce the impact of preventable disease for patients.
The Global Fund is a worldwide movement to defeat HIV, TB and malaria and ensure a healthier, safer, more equitable future for all. We raise and invest more than US$4 billion a year to fight the deadliest infectious diseases, challenge the injustice which fuels them and strengthen health systems in more than 100 of the hardest hit countries. We unite world leaders, communities, civil society, health workers and the private sector to find solutions that have the most impact, and we take them to scale worldwide. Since 2002, the Global Fund has saved 44 million lives. Information on the work of the Global Fund is available at www.theglobalfund.org.
Roche is a global pioneer in pharmaceuticals and diagnostics focused on advancing science to improve people’s lives. The combined strengths of pharmaceuticals and diagnostics under one roof have made Roche the leader in personalised healthcare – a strategy that aims to fit the right treatment to each patient in the best way possible.
Roche is the world’s largest biotech company, with truly differentiated medicines in oncology, immunology, infectious diseases, ophthalmology and diseases of the central nervous system. Roche is also the world leader in in vitro diagnostics and tissue-based cancer diagnostics, and a frontrunner in diabetes management.
Founded in 1896, Roche continues to search for better ways to prevent, diagnose and treat diseases and make a sustainable contribution to society. The company also aims to improve patient access to medical innovations by working with all relevant stakeholders. More than thirty medicines developed by Roche are included in the World Health Organization Model Lists of Essential Medicines, among them life-saving antibiotics, antimalarials and cancer medicines. Moreover, for the twelfth consecutive year, Roche has been recognised as one of the most sustainable companies in the Pharmaceuticals Industry by the Dow Jones Sustainability Indices (DJSI).
The Roche Group, headquartered in Basel, Switzerland, is active in over 100 countries and in 2020 employed more than 100,000 people worldwide. In 2020, Roche invested CHF 12.2 billion in R&D and posted sales of CHF 58.3 billion. Genentech, in the United States, is a wholly owned member of the Roche Group. Roche is the majority shareholder in Chugai Pharmaceutical, Japan
Acceptance based on two phase III studies that demonstrated early and sustained vision improvement with Vabysmo, meeting primary endpoint of non-inferiority compared to aflibercept
Application was further supported by data showing Vabysmo achieved rapid and robust drying of retinal fluid
If approved, RVO would be the third indication for Vabysmo in addition to neovascular or ‘wet’ age-related macular degeneration (nAMD) and diabetic macular edema (DME)
Vabysmo is currently approved in 60 countries to treat nAMD and DME, with nearly one million doses distributed globally
Basel, 9 May 2023 - Roche (SIX: RO, ROG; OTCQX: RHHBY) announced today that the U.S. Food and Drug Administration (FDA) has accepted the company’s supplemental Biologics License Application (sBLA) for Vabysmo® (faricimab) for the treatment of macular edema following retinal vein occlusion (RVO). The sBLA is based on results from the phase III BALATON and COMINO studies that demonstrated treatment with Vabysmo provided early and sustained improvement in vision, meeting the primary endpoint of non-inferior visual acuity gains at 24 weeks compared to aflibercept.1,2 Vabysmo’s safety profile was consistent with previous trials.
“This acceptance brings us one step closer to delivering Vabysmo as a treatment for retinal vein occlusion, a disease that affects more than one million people in the United States and can cause severe and sudden vision loss,” said Levi Garraway, M.D., Ph.D., Roche’s Chief Medical Officer and Head of Global Product Development. “If approved, this would be the third indication for Vabysmo, the first bispecific antibody available for the treatment of retinal conditions that can cause blindness.”
The data from the BALATON and COMINO studies will be submitted to other health authorities around the world, including the European Medicines Agency, for approval for the treatment of macular edema following RVO. The studies are ongoing, and data from weeks 24 to 72 will assess the potential of Vabysmo to extend dosing intervals up to every four months.1,2
Vabysmo is the first bispecific antibody approved for the eye and was approved in the United States for the treatment of neovascular or ‘wet’ age-related macular degeneration (nAMD) and diabetic macular edema (DME) in January 2022.3 Vabysmo has also been approved in 60 countries around the world, including Japan, the United Kingdom and in the European Union for people living with nAMD and DME.3-7 Neovascular AMD, DME and RVO together affect around 70 million people worldwide and are among the leading causes of vision loss.8-12
Vabysmo’s efficacy and safety profile in nAMD and DME is supported by four large, global studies involving more than 3,000 participants and extensive real-world experience, with nearly one million Vabysmo doses distributed globally.7,13-16 Vabysmo is the first bispecific antibody approved for the eye with phase III studies supporting treatment intervals of up to four months for people with these conditions.4 It targets and inhibits two signalling pathways linked to a number of vision-threatening retinal conditions by neutralising angiopoietin-2 (Ang-2) and vascular endothelial growth factor-A (VEGF-A).
BALATON (NCT04740905) and COMINO (NCT04740931) are two randomised, multicentre, double-masked, global phase III studies evaluating the efficacy and safety of Vabysmo® (faricimab) compared to aflibercept. For the first 20 weeks, patients are randomised 1:1 to receive six monthly injections of either Vabysmo (6.0 mg) or aflibercept (2.0 mg). From weeks 24 to 72, all patients receive Vabysmo (6.0 mg) up to every four months – according to a personalised treatment interval dosing regimen – using a treat-and-extend approach.
The BALATON study is being conducted in 553 patients with branch retinal vein occlusion. The COMINO study is being conducted in 729 patients with central retinal or hemiretinal vein occlusion.
The primary endpoint of each study is the change in best-corrected visual acuity from baseline at 24 weeks. Secondary endpoints include change in central subfield thickness (CST) from baseline over time up to 24 weeks.
Both studies met the primary endpoint, with Vabysmo showing non-inferior visual acuity gains compared to aflibercept. The average vision gains from baseline were comparable between the two treatments in both studies. In BALATON, vision gains were +16.9 eye chart letters in the Vabysmo arm and +17.5 letters in the aflibercept arm at 24 weeks. In COMINO, vision gains were +16.9 letters in the Vabysmo arm and +17.3 letters in the aflibercept arm at 24 weeks.
A secondary endpoint showed that Vabysmo achieved rapid and robust drying of retinal fluid, as measured by reduction in CST from baseline. In both studies, reductions in CST were comparable across treatment arms. In BALATON, CST reductions from baseline at 24 weeks were 311.4 μm in the Vabysmo arm and 304.4 μm in the aflibercept arm. In COMINO, CST reductions from baseline at 24 weeks were 461.6 μm in the Vabysmo arm and 448.8 μm in the aflibercept arm.
Additionally, both studies showed that more Vabysmo patients had an absence of blood vessel leakage in the retina compared to aflibercept patients as seen in a pre-specified exploratory endpoint. Blood vessel leakage in the macula may lead to more retinal fluid, which can cause swelling and blurry vision.
In BALATON, one-third of patients (34%) treated with Vabysmo had an absence of macular leakage compared to one-fifth (21%) of aflibercept patients at 24 weeks. In COMINO, the rates were 44% for Vabysmo patients versus 30% for aflibercept patients at 24 weeks.
In both studies, Vabysmo’s safety profile was consistent with previous trials. The most common adverse reaction was conjunctival haemorrhage (3%). Safety results were consistent across study arms.
The studies are ongoing, and data from weeks 24 to 72 will assess the potential of Vabysmo to extend dosing intervals up to every four months.
RVO is the second most common cause of vision loss due to retinal vascular diseases. It affects an estimated 28 million adults globally, mainly those aged 60 or older, and can lead to severe and sudden vision loss.8,17 RVO typically results in sudden, painless vision loss in the affected eye because the vein blockage restricts normal blood flow in the affected retina, resulting in ischaemia, bleeding, fluid leakage and retinal swelling called macular edema. Currently, macular edema due to RVO is typically treated with repeated intravitreal injections of anti-vascular endothelial growth factor therapies.19 There are two main types of RVO: branch RVO, which affects more than 23 million people globally and occurs when one of the four smaller ‘branches’ of the main central retinal vein becomes blocked; and central RVO, which is less common, affecting more than four million people worldwide, and occurs when the eye’s central retinal vein becomes blocked.
Roche has a robust phase III clinical development programme for Vabysmo. The programme includes AVONELLE-X, an extension study of TENAYA and LUCERNE evaluating the long-term safety and tolerability of Vabysmo in neovascular or ‘wet’ macular degeneration (nAMD), and Rhone-X, an extension study of YOSEMITE and RHINE evaluating the long-term safety and tolerability of Vabysmo in diabetic macular edema (DME).20,21 In addition, Roche is investigating the efficacy and safety of Vabysmo in people with macular edema following retinal vein occlusion (RVO) in two phase III studies, BALATON and COMINO.1,2 Roche has also initiated several phase IV studies, including the ELEVATUM study of Vabysmo in underrepresented patient populations with DME, the SALWEEN study of Vabysmo in a subpopulation of nAMD highly prevalent in Asia, as well as the VOYAGER study, a global real-world data collection platform.22-24 Roche also supports several other independent studies to further understand retinal conditions with a high unmet need.
Vabysmo is the first bispecific antibody approved for the eye.3,5 It targets and inhibits two signalling pathways linked to a number of vision-threatening retinal conditions by neutralising angiopoietin-2 (Ang-2) and vascular endothelial growth factor-A (VEGF-A). Ang-2 and VEGF-A contribute to vision loss by destabilising blood vessels, causing new leaky blood vessels to form and increasing inflammation.13,14 By blocking pathways involving Ang-2 and VEGF-A, Vabysmo is designed to stabilise blood vessels.13,14 Vabysmo is approved in 60 countries around the world, including the United States, Japan, the United Kingdom and in the European Union for people living with neovascular or ‘wet’ age-related macular degeneration and diabetic macular edema. Review by other regulatory authorities is ongoing.
Roche is focused on saving people’s eyesight from the leading causes of vision loss through pioneering therapies. Through our innovation in the scientific discovery of new potential drug targets, personalised healthcare, molecular engineering, biomarkers and continuous drug delivery, we strive to design the right therapies for the right patients.
We have the broadest retina pipeline in ophthalmology, which is led by science and informed by insights from people with eye diseases. Our pipeline includes gene therapies and treatments for geographic atrophy and other vision-threatening diseases, including rare and inherited conditions.
Applying our extensive experience, we have already brought breakthrough ophthalmic treatments to people living with vision loss. Susvimo™ (previously called Port Delivery System with ranibizumab) 100 mg/mL for intravitreal use via ocular implant is the first U.S. Food and Drug Administration-approved refillable eye implant for neovascular or ‘wet’ age-related macular degeneration that continuously delivers a customised formulation of ranibizumab over a period of months.25 Vabysmo® (faricimab) is the first bispecific antibody approved for the eye, which targets and inhibits two signalling pathways linked to a number of vision-threatening retinal conditions by neutralising angiopoietin-2 (Ang-2) and vascular endothelial growth factor-A (VEGF-A).3,5,13,14 Lucentis®* (ranibizumab injection) is the first treatment approved to improve vision in people with certain retinal conditions.
Roche is a global pioneer in pharmaceuticals and diagnostics focused on advancing science to improve people’s lives. The combined strengths of pharmaceuticals and diagnostics under one roof have made Roche the leader in personalised healthcare – a strategy that aims to fit the right treatment to each patient in the best way possible.
Roche is the world’s largest biotech company, with truly differentiated medicines in oncology, immunology, infectious diseases, ophthalmology and diseases of the central nervous system. Roche is also the world leader in in vitro diagnostics and tissue-based cancer diagnostics, and a frontrunner in diabetes management.
Founded in 1896, Roche continues to search for better ways to prevent, diagnose and treat diseases and make a sustainable contribution to society. The company also aims to improve patient access to medical innovations by working with all relevant stakeholders. More than thirty medicines developed by Roche are included in the World Health Organization Model Lists of Essential Medicines, among them life-saving antibiotics, antimalarials and cancer medicines. Moreover, for the twelfth consecutive year, Roche has been recognised as one of the most sustainable companies in the Pharmaceuticals Industry by the Dow Jones Sustainability Indices (DJSI).
The Roche Group, headquartered in Basel, Switzerland, is active in over 100 countries and in 2020 employed more than 100,000 people worldwide. In 2020, Roche invested CHF 12.2 billion in R&D and posted sales of CHF 58.3 billion. Genentech, in the United States, is a wholly owned member of the Roche Group. Roche is the majority shareholder in Chugai Pharmaceutical, Japan
Post-hoc analyses from four phase III studies indicate Vabysmo dried retinal fluid faster with fewer injections in neovascular or ‘wet’ age-related macular degeneration (nAMD) and diabetic macular edema (DME)
More Vabysmo patients with nAMD had absence of retinal fluid at 12 weeks in a post-hoc analysis from the phase III TENAYA and LUCERNE studies
DME patients treated with Vabysmo had less blood vessel leakage in the macula at 16 weeks in a post-hoc analysis from the phase III YOSEMITE and RHINE studies
Basel, 25 April 2023 - Roche (SIX: RO, ROG; OTCQX: RHHBY) announced today that post-hoc data indicate treatment with Vabysmo® (faricimab) led to greater and faster drying of retinal fluid with fewer injections compared to aflibercept in neovascular or ‘wet’ age-related macular degeneration (nAMD).1 In diabetic macular edema (DME), post-hoc data suggest Vabysmo treatment resulted in faster drying with fewer injections as well as less blood vessel leakage in the macula, the centre of the retina, compared to aflibercept.2,3 The analyses from the phase III TENAYA and LUCERNE (nAMD) and YOSEMITE and RHINE (DME) studies were shared at the 2023 Association for Research in Vision and Ophthalmology (ARVO) Annual Meeting, held from 23-27 April in New Orleans, United States.
“Reducing retinal fluid is associated with improved vision,” said Levi Garraway, M.D., Ph.D., Roche’s Chief Medical Officer and Head of Global Product Development. “These data continue to reinforce Vabysmo's ability to dry the retina and potential to make a meaningful difference for people with vision-threatening eye conditions.”
Vabysmo is the first bispecific antibody for the eye and is currently approved in 60 countries to treat nAMD and DME, with more than 800,000 Vabysmo doses distributed globally.4 Neovascular AMD and DME are two of the leading causes of vision loss worldwide, affecting more than 40 million people.5-8 In these conditions, blood vessel leakage can cause a build-up of fluid and swelling in the back of the eye, contributing to sight loss.9,10
“These findings suggest that Vabysmo may provide better stability of blood vessels in the macula,” said Roger Goldberg, M.D., MBA, an ophthalmologist at Bay Area Retina Associates in Walnut Creek, California, United States, and a Vabysmo phase III study investigator. “Blood vessel stability may contribute to faster drying and extended durability.”
A post-hoc analysis of pooled data from the head-to-head dosing period (weeks 0-12) of the phase III TENAYA and LUCERNE studies in nAMD showed:*
Vabysmo reduced retinal fluid from baseline compared to aflibercept, as measured by reduction in central subfield thickness (CST).
At 12 weeks, CST reductions were 145 µm in the Vabysmo arm and 133 µm in the aflibercept arm.
A larger proportion of Vabysmo patients (77%) had absence of retinal fluid at 12 weeks versus aflibercept (67%), as measured by subretinal and intraretinal fluid (SRF and IRF).
Absence of retinal fluid, as measured by absence of SRF and IRF observed in 75% of patients in each treatment arm, occurred at eight weeks with Vabysmo versus 12 weeks with aflibercept, corresponding to a fewer number of injections for Vabysmo patients versus aflibercept.
A post-hoc analysis of pooled two-year data from the phase III YOSEMITE and RHINE studies in DME compared time to fluid control between Vabysmo and aflibercept, as measured by absence of DME and absence of IRF. The analysis showed:*
Absence of DME, defined as CST <325 µm observed in 75% of patients in each treatment arm, occurred at 20 weeks with Vabysmo versus 36 weeks with aflibercept – a difference of nearly four months.
Absence of retinal fluid, as measured by absence of IRF observed in 50% of patients in each treatment arm, occurred more than eight months earlier in Vabysmo patients versus aflibercept.
Absence of IRF occurred at 48 weeks with Vabysmo versus 84 weeks with aflibercept, corresponding to a fewer number of injections for Vabysmo patients versus aflibercept.
A separate post-hoc analysis of pooled data from the head-to-head dosing period (weeks 0-16) of the YOSEMITE and RHINE studies evaluated blood vessel leakage in the macula – an important marker of vascular stability. Blood vessel leakage in the macula may lead to more retinal fluid, which can cause swelling and blurry vision.11 Results showed:*
The macular leakage area in Vabysmo patients was more than 50% smaller compared to aflibercept at 16 weeks.
Vabysmo reduced the macular leakage area to 3.6 mm2 from baseline compared to 7.6 mm2 with aflibercept.
Nearly twice as many patients (28.4%) had resolution of leakage versus aflibercept (15.2%) at 16 weeks.
TENAYA (NCT03823287) and LUCERNE (NCT03823300) are two identical, randomised, multicentre, double-masked, global phase III studies evaluating the efficacy and safety of Vabysmo® (faricimab) compared to aflibercept in 1,329 people living with neovascular or ‘wet’ age-related macular degeneration (671 in TENAYA and 658 in LUCERNE). The studies each have two treatment arms: Vabysmo 6.0 mg administered at intervals of two, three or four months, following four initial monthly doses, selected based on objective assessment of disease activity as measured by optical coherence tomography and visual acuity evaluations at weeks 20 and 24; and aflibercept 2.0 mg administered at fixed two-month intervals after three initial monthly doses. At week 60, patients randomised to the Vabysmo arm were treated using a treat-and-extend approach up to week 108. Dosing schedule for Vabysmo patients during the treat-and-extend phase was adjusted based on treatment response as determined by central subfield thickness (CST) and visual acuity. In both arms, placebo injections were administered at study visits when treatment injections were not scheduled to maintain the masking of investigators and participants.
The primary endpoint of the studies is the average change in best-corrected visual acuity (BCVA) score (the best distance vision a person can achieve – including with correction such as glasses – when reading letters on an eye chart) from baseline, averaged over weeks 40, 44 and 48. Secondary endpoints include safety; the percentage of participants in the Vabysmo arm receiving treatment every two, three and four months; the percentage of participants achieving a gain, and the percentage avoiding a loss, of 15 letters or more in BCVA from baseline over time; and change in CST from baseline over time.
YOSEMITE (NCT03622580) and RHINE (NCT03622593) are two identical, randomised, multicentre, double-masked, global phase III studies evaluating the efficacy and safety of Vabysmo® (faricimab) compared to aflibercept in 1,891 people with diabetic macular edema (940 in YOSEMITE and 951 in RHINE). The studies each have three treatment arms: Vabysmo 6.0 mg administered up to every four months after four initial monthly doses using a treat-and-extend approach; Vabysmo 6.0 mg administered at two-month intervals after six initial monthly doses; and aflibercept 2.0 mg administered at fixed two-month intervals after five initial monthly doses. Dosing schedule for patients within the treat-and-extend arm was determined by central subfield thickness (CST) and visual acuity. In all three arms, placebo injections were administered at study visits when treatment injections were not scheduled to maintain the masking of investigators and participants.
The primary endpoint of the studies is the average change in best-corrected visual acuity (BCVA) score (the best distance vision a person can achieve – including with correction such as glasses – when reading letters on an eye chart) from baseline at one year, averaged over weeks 48, 52 and 56. Secondary endpoints include: safety; the percentage of participants in the treat-and-extend arm receiving Vabysmo every one, two, three and four months, at week 52; the percentage of participants achieving a two-step or greater improvement from baseline in diabetic retinopathy severity at week 52; the percentage of participants achieving a gain, and the percentage avoiding a loss, of 15 letters or more in BCVA from baseline over time; change in CST from baseline over time; and percentage of patients with absence of intraretinal fluid over time.
Age-related macular degeneration (AMD) is a condition that affects the part of the eye that provides sharp, central vision needed for activities like reading.4 Neovascular or ‘wet’ AMD (nAMD) is an advanced form of the disease that can cause rapid and severe vision loss if left untreated.16,17 It develops when new and abnormal blood vessels grow uncontrolled under the macula, causing swelling, bleeding and/or fibrosis.16 Worldwide, around 20 million people are living with nAMD – the leading cause of vision loss in people over the age of 60 – and the condition will affect even more people around the world as the global population ages.
Affecting around 21 million people globally, diabetic macular edema (DME) is a vision-threatening retinal condition associated with blindness and decreased quality of life when left untreated.7 DME occurs when damaged blood vessels leak into and cause swelling in the macula – the central area of the retina responsible for the sharp vision needed for reading and driving.9,11 The number of people with DME is expected to grow as the prevalence of diabetes increases.
Roche has a robust phase III clinical development programme for Vabysmo® (faricimab). The programme includes AVONELLE-X, an extension study of TENAYA and LUCERNE evaluating the long-term safety and tolerability of Vabysmo in neovascular or ‘wet’ age-related macular degeneration (nAMD), and Rhone-X, an extension study of YOSEMITE and RHINE evaluating the long-term safety and tolerability of Vabysmo in diabetic macular edema (DME).19,20 In addition, Roche is investigating the efficacy and safety of Vabysmo in people with macular edema following retinal vein occlusion in two phase III studies, BALATON and COMINO.21,22 Roche has also initiated several phase IV studies, including the Elevatum study of Vabysmo in underrepresented patient populations with DME, the SALWEEN study of Vabysmo in a subpopulation of nAMD highly prevalent in Asia, as well as the VOYAGER study, a global real-world data collection platform.23-25 Roche also supports several other independent studies to further understand retinal conditions with a high unmet need.
Vabysmo® (faricimab) is the first bispecific antibody approved for the eye.26,27 It targets and inhibits two signaling pathways linked to a number of vision-threatening retinal conditions by neutralising angiopoietin-2 (Ang-2) and vascular endothelial growth factor-A (VEGF-A).28,29 Ang-2 and VEGF-A contribute to vision loss by destabilising blood vessels, causing new leaky blood vessels to form and increasing inflammation.28,29 By blocking pathways involving Ang-2 and VEGF-A, Vabysmo is designed to stabilise blood vessels.28,29 Vabysmo is approved in 60 countries around the world, including the United States, Japan, the United Kingdom and in the European Union for people living with neovascular or ‘wet’ age-related macular degeneration and diabetic macular edema.
Roche is focused on saving people’s eyesight from the leading causes of vision loss through pioneering therapies. Through our innovation in the scientific discovery of new potential drug targets, personalised healthcare, molecular engineering, biomarkers and continuous drug delivery, we strive to design the right therapies for the right patients.
We have the broadest retina pipeline in ophthalmology, which is led by science and informed by insights from people with eye diseases. Our pipeline includes gene therapies and treatments for geographic atrophy and other vision-threatening diseases, including rare and inherited conditions.
Applying our extensive experience, we have already brought breakthrough ophthalmic treatments to people living with vision loss. Susvimo™ (Port Delivery System with ranibizumab) 100 mg/mL for intravitreal use via ocular implant is the first U.S. Food and Drug Administration-approved refillable eye implant for neovascular or ‘wet’ age-related macular degeneration that continuously delivers a customised formulation of ranibizumab over a period of months.32 Vabysmo® (faricimab) is the first bispecific antibody approved for the eye, which targets and inhibits two signaling pathways linked to a number of vision-threatening retinal conditions by neutralising angiopoietin-2 (Ang-2) and vascular endothelial growth factor-A (VEGF-A).26-29 Lucentis®** (ranibizumab injection) is the first treatment approved to improve vision in people with certain retinal conditions.
Roche is a global pioneer in pharmaceuticals and diagnostics focused on advancing science to improve people’s lives. The combined strengths of pharmaceuticals and diagnostics under one roof have made Roche the leader in personalised healthcare – a strategy that aims to fit the right treatment to each patient in the best way possible.
Roche is the world’s largest biotech company, with truly differentiated medicines in oncology, immunology, infectious diseases, ophthalmology and diseases of the central nervous system. Roche is also the world leader in in vitro diagnostics and tissue-based cancer diagnostics, and a frontrunner in diabetes management.
Founded in 1896, Roche continues to search for better ways to prevent, diagnose and treat diseases and make a sustainable contribution to society. The company also aims to improve patient access to medical innovations by working with all relevant stakeholders. More than thirty medicines developed by Roche are included in the World Health Organization Model Lists of Essential Medicines, among them life-saving antibiotics, antimalarials and cancer medicines. Moreover, for the twelfth consecutive year, Roche has been recognised as one of the most sustainable companies in the Pharmaceuticals Industry by the Dow Jones Sustainability Indices (DJSI).
The Roche Group, headquartered in Basel, Switzerland, is active in over 100 countries and in 2020 employed more than 100,000 people worldwide. In 2020, Roche invested CHF 12.2 billion in R&D and posted sales of CHF 58.3 billion. Genentech, in the United States, is a wholly owned member of the Roche Group. Roche is the majority shareholder in Chugai Pharmaceutical, Japan
Polivy combination is the first FDA-approved therapy in nearly 20 years for the first-line treatment of diffuse large B-cell lymphoma, an aggressive disease and the most common form of non-Hodgkin lymphoma in the US
POLARIX trial showed the Polivy combination reduced the risk of disease progression, relapse or death by 27% compared to the standard of care, R-CHOP, with a comparable safety profile
First-line treatment with Polivy plus R-CHP has the potential to reduce the burden on patients and healthcare systems, associated with disease progression
Basel, 19 April 2023 - Roche (SIX: RO, ROG; OTCQX: RHHBY) announced today that the U.S. Food and Drug Administration (FDA) has approved Polivy® (polatuzumab vedotin-piiq) in combination with Rituxan® (rituximab), cyclophosphamide, doxorubicin and prednisone (R-CHP) for the treatment of adult patients who have previously untreated diffuse large B-cell lymphoma (DLBCL), not otherwise specified (NOS) or high-grade B-cell lymphoma (HGBL) and who have an International Prognostic Index (IPI) score of two or greater. This FDA decision converts the accelerated approval of Polivy in combination with bendamustine and Rituxan for relapsed or refractory (R/R) DLBCL after at least two prior therapies to regular approval.
DLBCL is an aggressive, hard-to-treat disease and is the most common form of non-Hodgkin lymphoma in the United States. Approximately 31,000 people in the US are projected to be diagnosed with DLBCL in 2023. Limited progress has been made in improving patient outcomes in previously untreated DLBCL over the last two decades. While many patients are responsive to initial treatment, as many as four in 10 people with DLBCL do not respond or relapse. For people who undergo initial treatment with the standard of care, MabThera/Rituxan plus cyclophosphamide, doxorubicin, vincristine and prednisone (R-CHOP), most relapses occur within two years of starting treatment, and the majority of those who require subsequent lines of therapy have poor outcomes.
“It has been nearly 20 years since a new treatment option has become available to people newly diagnosed with diffuse large B-cell lymphoma,” said Levi Garraway, M.D., Ph.D., Roche’s Chief Medical Officer and Head of Global Product Development. “Today’s decision from the FDA to approve Polivy in combination with R-CHP in this setting brings a much-needed new treatment option which may improve outcomes and bring other benefits to many patients with this aggressive lymphoma.”
The FDA approval of Polivy plus R-CHP for the first-line treatment of DLBCL is based on pivotal data from POLARIX, an international phase III, randomised, double-blind, placebo-controlled study that demonstrated a statistically significant and clinically meaningful improvement in PFS compared to R-CHOP. The risk of disease progression, relapse or death was reduced by 27% with Polivy plus R-CHP (n=440) compared with R-CHOP (n=439; hazard ratio [HR] 0.73; 95% confidence interval [CI]: 0.57–0.95; p<0.02). The safety profile was comparable for Polivy plus R-CHP versus R-CHOP, including rates of Grade 3-4 adverse events (AEs; 57.7% versus 57.5%), serious AEs (34.0% versus 30.6%), Grade 5 AEs (3.0% versus 2.3%), and AEs leading to dose reduction (9.2% versus 13.0%). The most common AEs were peripheral neuropathy, nausea, fatigue, diarrhoea, constipation, alopecia, and mucositis. The most common Grade 3-4 AEs were lymphopenia and neutropenia.
This approval follows the FDA Oncologic Drugs Advisory Committee (ODAC) vote of 11 to 2 in favour of Polivy in combination with R-CHP for previously untreated DLBCL. More than 70 countries have approved this Polivy combination for the treatment of adult patients with previously untreated DLBCL, including in the EU, UK, Japan, Canada and China. Polivy in combination with R-CHP was recently added to the National Comprehensive Cancer Network® (NCCN®) Clinical Practice Guidelines in Oncology (NCCN Guidelines®) as a category 1, preferred regimen for first-line DLBCL. Polivy in combination with bendamustine and MabThera/Rituxan is currently approved in more than 80 countries worldwide for the treatment of adults with relapsed or refractory (R/R) DLBCL after one or more prior therapies, including in the US.
Roche continues to explore areas of unmet need where Polivy has the potential to deliver additional benefit, including in ongoing studies investigating combinations of Polivy with the company’s CD20xCD3 T-cell engaging bispecific antibodies Lunsumio® (mosunetuzumab) or Columvi® (glofitamab). Trials include the phase III SUNMO study in combination with Lunsumio in patients with R/R DLBCL and the phase III POLARGO study with MabThera/Rituxan in combination with gemcitabine and oxaliplatin in patients with R/R DLBCL.
POLARIX [NCT03274492] is an international phase III, randomised, double-blind, placebo-controlled study evaluating the efficacy, safety and pharmacokinetics of Polivy® (polatuzumab vedotin) plus MabThera®/Rituxan® (rituximab), cyclophosphamide, doxorubicin and prednisone (R-CHP) versus MabThera/Rituxan, cyclophosphamide, doxorubicin, vincristine and prednisone (R-CHOP) in people with previously untreated diffuse large B-cell lymphoma (DLBCL). Eight-hundred and seventy-nine patients were randomised 1:1 to receive either Polivy plus R-CHP plus a vincristine placebo for six cycles, followed by MabThera/Rituxan for two cycles; or R-CHOP plus a Polivy placebo for six cycles, followed by two cycles of MabThera/Rituxan. The primary outcome measure is progression-free survival as assessed by the investigator using the Lugano Response Criteria for malignant lymphoma. POLARIX is being conducted in collaboration with The Lymphoma Study Association and The Lymphoma Academic Research Organisation.
Diffuse large B-cell lymphoma (DLBCL) is the most common form of non-Hodgkin lymphoma (NHL), accounting for about one in three cases of NHL.1 DLBCL is an aggressive (fast-growing) type of NHL.1 While it is generally responsive to treatment in the frontline, as many as 40% of people will relapse or have refractory disease, at which time salvage therapy options are limited and survival is short.2,3 Approximately 160,000 people worldwide are estimated to be diagnosed with DLBCL each year.
Polivy is a first-in-class anti-CD79b antibody-drug conjugate (ADC). The CD79b protein is expressed specifically in the majority of B-cells, an immune cell impacted in some types of non-Hodgkin lymphoma (NHL), making it a promising target for the development of new therapies. Polivy binds to cancer cells such as CD79b and destroys these B-cells through the delivery of an anti-cancer agent, which is thought to minimise the effects on normal cells. Polivy is being developed by Roche using Seagen ADC technology and is currently being investigated for the treatment of several types of NHL.
Roche has been developing medicines for people with malignant and non-malignant blood diseases for more than 20 years; our experience and knowledge in this therapeutic area runs deep. Today, we are investing more than ever in our effort to bring innovative treatment options to patients across a wide range of haematologic diseases. Our approved medicines include MabThera®/Rituxan® (rituximab), Gazyva®/Gazyvaro® (obinutuzumab), Polivy® (polatuzumab vedotin-piiq), Venclexta®/Venclyxto® (venetoclax) in collaboration with AbbVie, Hemlibra® (emicizumab), Lunsumio® (mosunetuzumab) and Columvi® (glofitamab). Our pipeline of investigational haematology medicines includes a T-cell engaging bispecific antibody cevostamab, targeting both FcRH5 and CD3; Tecentriq® (atezolizumab), a monoclonal antibody designed to bind with PD-L1 and crovalimab, an anti-C5 antibody engineered to optimise complement inhibition. Our scientific expertise, combined with the breadth of our portfolio and pipeline, also provides a unique opportunity to develop combination regimens that aim to improve the lives of patients even further.
Roche is a global pioneer in pharmaceuticals and diagnostics focused on advancing science to improve people’s lives. The combined strengths of pharmaceuticals and diagnostics under one roof have made Roche the leader in personalised healthcare – a strategy that aims to fit the right treatment to each patient in the best way possible.
Roche is the world’s largest biotech company, with truly differentiated medicines in oncology, immunology, infectious diseases, ophthalmology and diseases of the central nervous system. Roche is also the world leader in in vitro diagnostics and tissue-based cancer diagnostics, and a frontrunner in diabetes management.
Founded in 1896, Roche continues to search for better ways to prevent, diagnose and treat diseases and make a sustainable contribution to society. The company also aims to improve patient access to medical innovations by working with all relevant stakeholders. More than thirty medicines developed by Roche are included in the World Health Organization Model Lists of Essential Medicines, among them life-saving antibiotics, antimalarials and cancer medicines. Moreover, for the twelfth consecutive year, Roche has been recognised as one of the most sustainable companies in the Pharmaceuticals Industry by the Dow Jones Sustainability Indices (DJSI).
The Roche Group, headquartered in Basel, Switzerland, is active in over 100 countries and in 2020 employed more than 100,000 people worldwide. In 2020, Roche invested CHF 12.2 billion in R&D and posted sales of CHF 58.3 billion. Genentech, in the United States, is a wholly owned member of the Roche Group. Roche is the majority shareholder in Chugai Pharmaceutical, Japan
In the first-ever positive Phase III trial in the adjuvant hepatocellular carcinoma (HCC) setting, Tecentriq plus Avastin reduced the risk of disease recurrence by 28%
Up to 80% of people with this type of HCC experience disease recurrence, at which point they are faced with poorer prognosis and shorter survival
These data will be presented at the American Association for Cancer Research (AACR) Annual Meeting 2023 and included in the official press programme
Basel, 16 April 2023 - Roche (SIX: RO, ROG; OTCQX: RHHBY) announced today new data from the Phase III IMbrave050 study that show Tecentriq® (atezolizumab) plus Avastin® (bevacizumab) demonstrated a statistically significant improvement in recurrence-free survival (RFS) in people with hepatocellular carcinoma (HCC) at high risk of disease recurrence following liver resection or ablation with curative intent.2
“Four out of five people with HCC who receive surgery with curative intent may still see their cancer return. Thus, an urgent need exists for adjuvant treatments to prevent early recurrence and improve survival rates,” said Levi Garraway, M.D., Ph.D., Chief Medical Officer and Head of Global Product Development. “With Tecentriq plus Avastin already a standard of care in unresectable HCC, we are pleased with the potential of these results and look forward to seeing more mature data.”
The Tecentriq investigational combination reduced the risk of cancer returning by 28%, compared with active surveillance, at a median follow-up of 17.4 months (independent review facility [IRF]-RFS hazard ratio [HR]=0.72, 95% CI: 0.56-0.93; P=0.0120).1 The IRF-RFS findings were generally consistent across clinical subgroups. Overall Survival (OS), a key secondary endpoint, was immature (7% event-rate) at the time of data analysis. The safety data for Tecentriq plus Avastin were consistent with the well-established safety profile of each therapeutic treatment and with the underlying disease.1
The late-breaking data will be presented at the AACR Annual Meeting 2023 and have been included as part of the official press programme. Discussions with health authorities are ongoing and follow-up will continue for the final RFS data and more mature OS data at the next planned analysis.
The IMbrave050 study is part of Roche’s overall commitment to drive fundamental treatment change and improve outcomes for people living with liver cancer. Tecentriq plus Avastin was the first treatment in over a decade to significantly improve OS over the existing standard of care, based on data from the IMbrave150 study.3 The Tecentriq combination quickly became a standard of care in unresectable HCC and is clearly defined as a preferred front-line treatment in multiple international clinical guidelines.
IMbrave050 is a Phase III global, multicentre, open-label, randomised study evaluating the efficacy and safety of adjuvant Tecentriq plus Avastin, compared with active surveillance, in people with HCC at high risk of recurrence (determined by the size and number of cancerous lesions and the histopathology results, if available) after surgical resection or ablation with curative intent.
The study randomised 668 people with a ratio of 1:1 to receive either Tecentriq (1,200 mg every three weeks) plus Avastin (15 mg/kg every three weeks) for a period of 12 months or 17 cycles, or no intervention with active surveillance. The primary endpoint is independent review facility-assessed RFS. Key secondary endpoints include OS, RFS as determined by the investigator and RFS in patients with PD-L1-positive disease.
Liver cancer is the third leading cause of cancer death and one of the few cancers where mortality is rising.4,5 More than 900,000 people are diagnosed with the disease globally each year, which translates to one person diagnosed every 90 seconds.4 Nine out of ten cases of HCC are caused by chronic liver disease, which includes chronic hepatitis B and C infection, non-alcoholic fatty liver disease (NAFLD), non-alcoholic steatohepatitis (NASH), alcohol-related liver disease (ALD) and cirrhosis resulting from these conditions.5,6
If diagnosed in the early stage, surgery may be prescribed to remove the primary tumour, however an estimated 70-80% of people with early-stage HCC experience disease recurrence following surgery.2 Early recurrence is associated with poorer prognosis and shorter survival.2,7 Tumour size, number of tumours, and portal vein invasion are associated with an increased risk of recurrence.
Tecentriq is a cancer immunotherapy approved for some of the most aggressive and difficult-to-treat forms of cancer. Tecentriq was the first cancer immunotherapy approved for the treatment of a certain type of early-stage non-small cell lung cancer (NSCLC), small cell lung cancer (SCLC) and HCC. Tecentriq is also approved in countries around the world, either alone or in combination with targeted therapies and/or chemotherapies, for various forms of metastatic NSCLC, certain types of metastatic urothelial cancer, PD-L1-positive metastatic triple-negative breast cancer and BRAF V600 mutation-positive advanced melanoma.
Tecentriq is a monoclonal antibody designed to bind with a protein called programmed death ligand-1 (PD-L1), which is expressed on tumour cells and tumour-infiltrating immune cells, blocking its interactions with both PD-1 and B7.1 receptors. By inhibiting PD-L1, Tecentriq may enable the activation of T-cells. Tecentriq is a cancer immunotherapy that has the potential to be used as a foundational combination partner with other immunotherapies, targeted medicines and various chemotherapies across a broad range of cancers. In addition to intravenous infusion, the formulation of Tecentriq is also being investigated as a subcutaneous injection to help address the growing burden of cancer treatment for patients and healthcare systems.
Roche is a global pioneer in pharmaceuticals and diagnostics focused on advancing science to improve people’s lives. The combined strengths of pharmaceuticals and diagnostics under one roof have made Roche the leader in personalised healthcare – a strategy that aims to fit the right treatment to each patient in the best way possible.
Roche is the world’s largest biotech company, with truly differentiated medicines in oncology, immunology, infectious diseases, ophthalmology and diseases of the central nervous system. Roche is also the world leader in in vitro diagnostics and tissue-based cancer diagnostics, and a frontrunner in diabetes management.
Founded in 1896, Roche continues to search for better ways to prevent, diagnose and treat diseases and make a sustainable contribution to society. The company also aims to improve patient access to medical innovations by working with all relevant stakeholders. More than thirty medicines developed by Roche are included in the World Health Organization Model Lists of Essential Medicines, among them life-saving antibiotics, antimalarials and cancer medicines. Moreover, for the twelfth consecutive year, Roche has been recognised as one of the most sustainable companies in the Pharmaceuticals Industry by the Dow Jones Sustainability Indices (DJSI).
The Roche Group, headquartered in Basel, Switzerland, is active in over 100 countries and in 2020 employed more than 100,000 people worldwide. In 2020, Roche invested CHF 12.2 billion in R&D and posted sales of CHF 58.3 billion. Genentech, in the United States, is a wholly owned member of the Roche Group. Roche is the majority shareholder in Chugai Pharmaceutical, Japan
Vabysmo data suggest rapid and robust drying of retinal fluid in patients with neovascular or ‘wet’ age-related macular degeneration and diabetic macular edema
Real-world studies of Vabysmo demonstrate ability to extend treatment intervals in the first four months while maintaining visual acuity
Clinical data on an investigational anti-interleukin-6 treatment in uveitic macular edema will be presented for the first time
Basel, 13 April 2023 - Roche (SIX: RO, ROG; OTCQX: RHHBY) announced today that new data for its approved and investigational medicines will be highlighted in 30 abstracts at the 2023 Association for Research in Vision and Ophthalmology (ARVO) Annual Meeting, which will be held from 23-27 April 2023 in New Orleans, United States. The abstracts showcase the strength and breadth of Roche’s Ophthalmology portfolio, including post-hoc data from phase III Vabysmo® (faricimab) studies that support its benefit in drying retinal fluid in neovascular or ‘wet’ age-related macular degeneration (nAMD) and diabetic macular edema (DME).1-3 Real-world data on Vabysmo treatment patterns and outcomes will be presented, as well as approaches to personalised healthcare that include the use of artificial intelligence (AI) modelling to predict retinal disease progression.4-7 Additionally, phase I data for an investigational anti-interleukin-6 (IL-6) treatment in uveitic macular edema (UME), to be presented for the first time, suggest the monoclonal antibody may improve visual acuity in patients with UME.8
“The breadth of data we are presenting at ARVO demonstrates our sustained commitment to preserving vision for people with potentially blinding retinal conditions,” said Levi Garraway, M.D., Ph.D., Chief Medical Officer and Head of Global Product Development. “We are particularly encouraged by data indicating that Vabysmo may stabilise blood vessels and reduce fluid in the retina. Fluid control is essential for optimal central vision used for everyday activities such as reading and driving.”
The following data will be presented at ARVO 2023:
A post-hoc analysis from the head-to-head dosing period of the phase III TENAYA and LUCERNE studies suggests Vabysmo results in greater drying of retinal fluid compared to aflibercept in people with nAMD. The data include change in central subfield thickness (CST), absence of subretinal and intraretinal fluid (SRF and IRF) and time to absence of SRF and IRF.
A post-hoc analysis from the head-to-head dosing period of the phase III YOSEMITE and RHINE studies also supports the positive impact of Vabysmo on macular blood vessel leakage compared to aflibercept in people with DME. Outcomes include macular leakage area and the proportion of patients with minimal to no macular leakage - two important markers of vascular stability. Another analysis from YOSEMITE and RHINE suggests Vabysmo reduces retinal fluid when compared to aflibercept in people with DME. The data include time to absence of DME (CST <325 µm) and time to absence of IRF.
Two Vabysmo real-world studies in nAMD and DME show patients extended their dosing intervals early in their treatment while maintaining or improving their vision. The majority of patients were able to extend their treatment intervals during the four initial doses. Treatment intervals were categorised as ‘extended’ if any interval was more than six weeks apart.
Phase I data on an IL-6 inhibitor that is in development for UME and other retinal conditions suggest that this investigational monoclonal antibody improves visual acuity and CST in patients with UME.
The IL-6 pathway plays an important role in the development and progression of UME by promoting blood vessel leakage and inflammation.9 UME is a complication of uveitis, a form of eye inflammation.10 This results in accumulation of fluid in the macula and can lead to significant visual impairment and vision loss.10 The estimated prevalence of uveitis is between 6 to 12 people per 10,000 globally, and approximately one-third of these people are impacted by UME.11
Roche recently launched two phase III trials in UME based on encouraging phase I safety and efficacy data. The first patients have been treated in the Meerkat (NCT05642312) and Sandcat (NCT05642325) studies, which are evaluating the safety and efficacy of the monoclonal antibody in people with UME.12,13 Roche is also studying the IL-6 inhibitor in DME.
Roche will also present research related to the diagnosis and treatment of retinal conditions. These presentations include new research on the use of AI and machine learning to predict disease progression in geographic atrophy, a progressive and irreversible form of AMD; enable timely and accurate assessment of disease activity in nAMD or DME; predict treatment response in DME; and investigate new imaging biomarkers in diabetic retinopathy.
Further information on select Roche abstracts that will be presented at ARVO 2023 can be found in the table below.
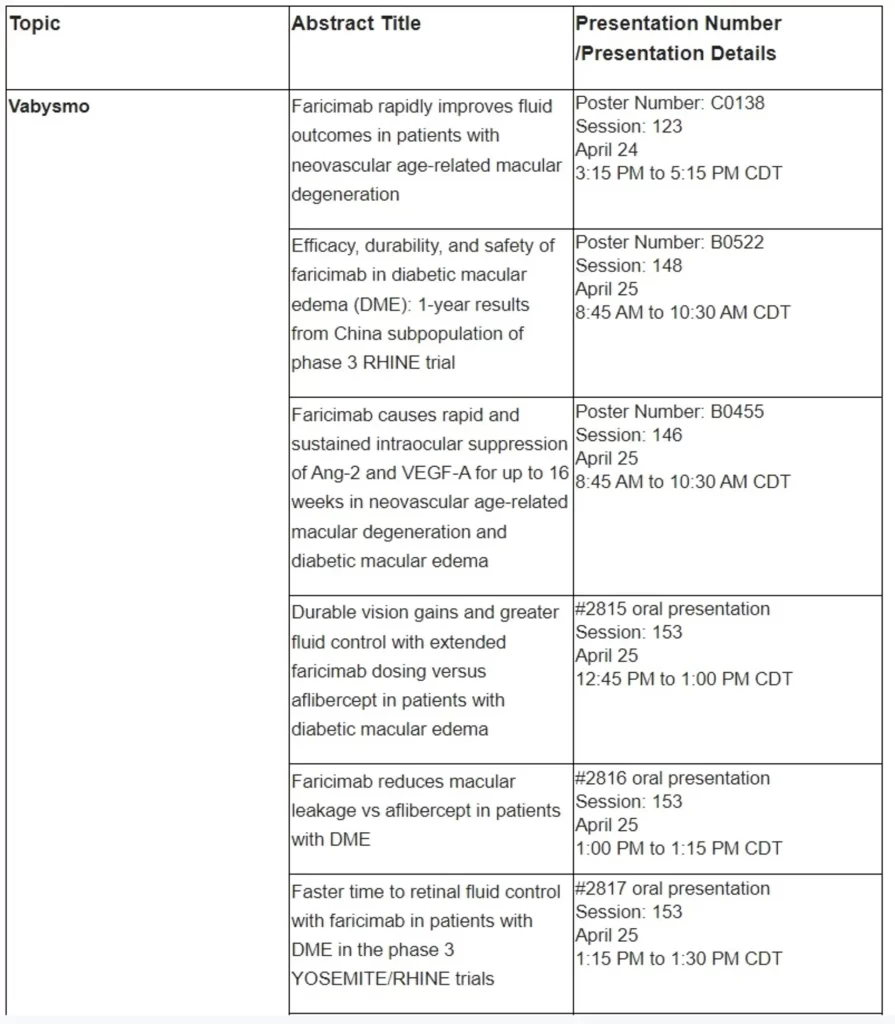
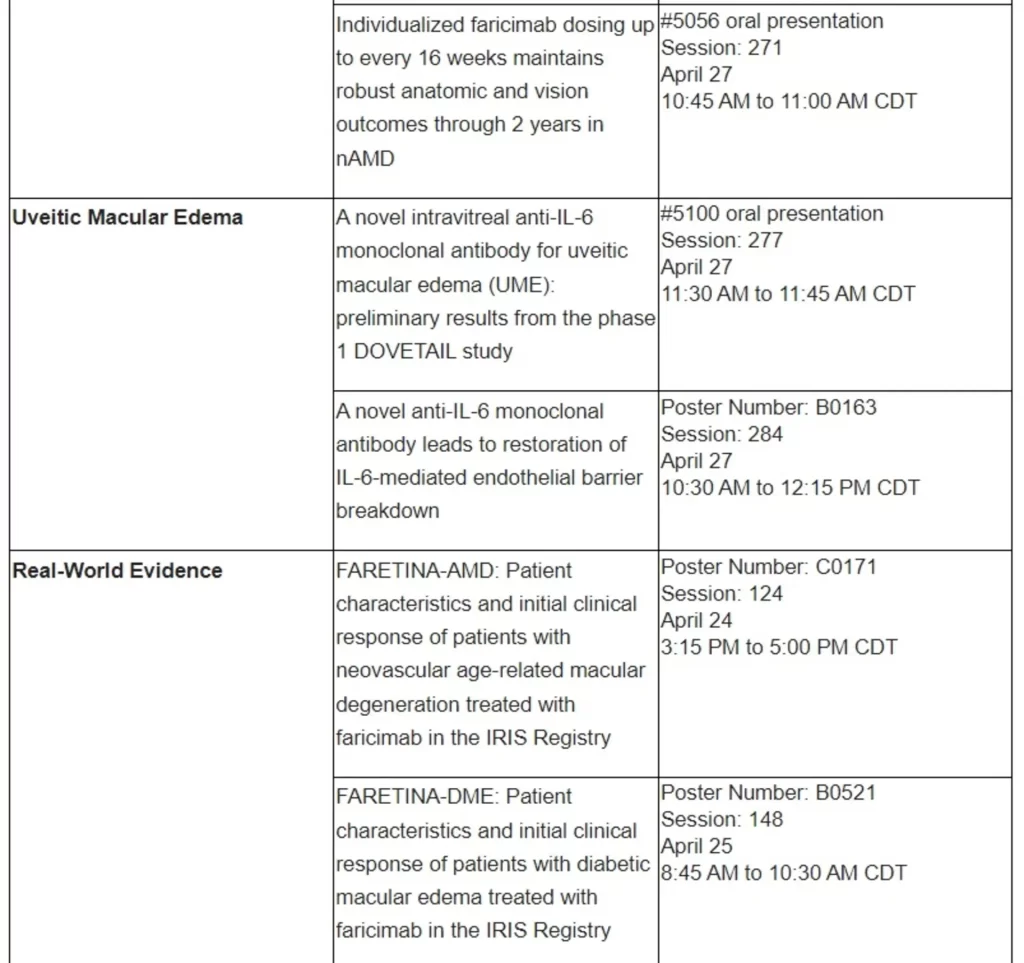
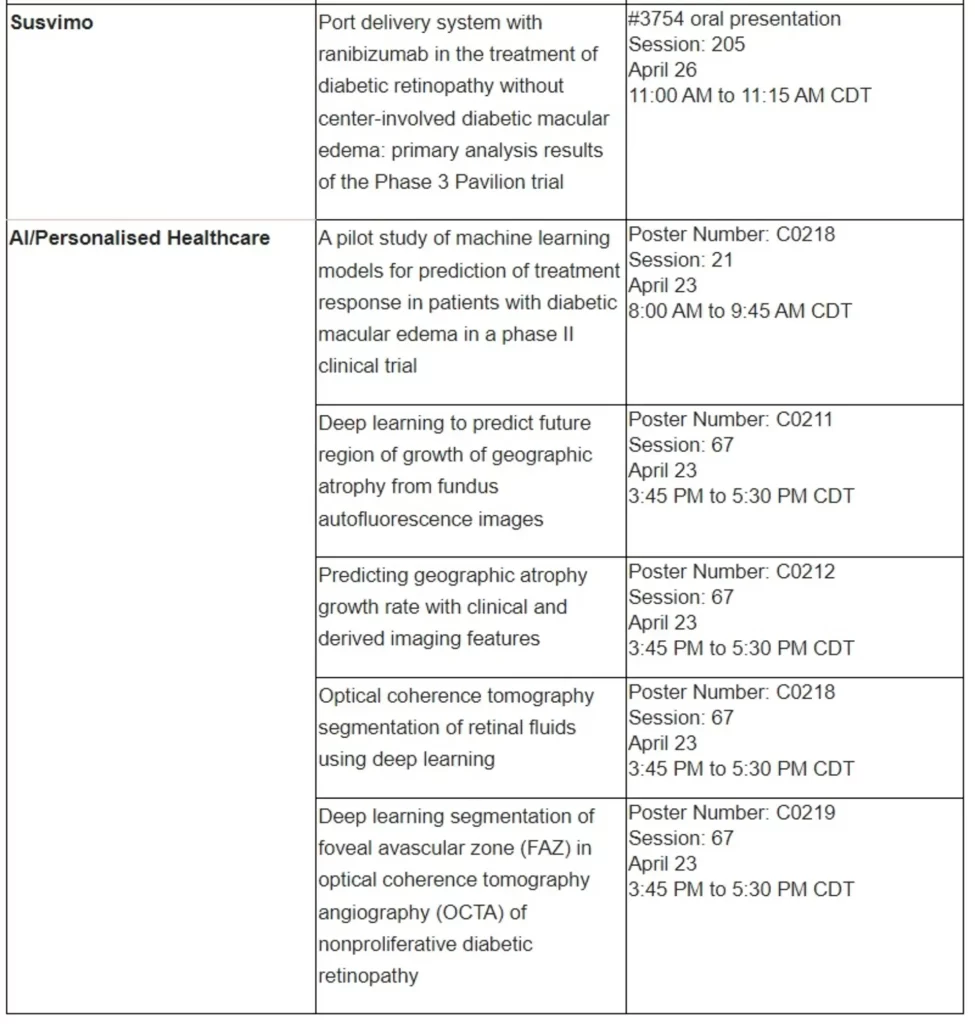
Age-related macular degeneration (AMD) is a condition that affects the part of the eye that provides sharp, central vision needed for activities like reading.18 Neovascular or ‘wet’ AMD (nAMD) is an advanced form of the disease that can cause rapid and severe vision loss if left untreated.19,20 It develops when new and abnormal blood vessels grow uncontrolled under the macula, causing swelling, bleeding and/or fibrosis.19 Worldwide, around 20 million people are living with nAMD – the leading cause of vision loss in people over the age of 60 – and the condition will affect even more people around the world as the global population ages.
Affecting around 21 million people globally, diabetic macular edema (DME) is a vision-threatening retinal condition associated with blindness and decreased quality of life when left untreated.23 DME occurs when damaged blood vessels leak into and cause swelling in the macula – the central area of the retina responsible for the sharp vision needed for reading and driving.24,25 The number of people with DME is expected to grow as the prevalence of diabetes increases.
Roche has a robust phase III clinical development programme for Vabysmo. The programme includes AVONELLE-X, an extension study of TENAYA and LUCERNE evaluating the long-term safety and tolerability of Vabysmo in neovascular or ‘wet’ age-related macular degeneration (nAMD), and Rhone-X, an extension study of YOSEMITE and RHINE evaluating the long-term safety and tolerability of Vabysmo in diabetic macular edema (DME).27,28 In addition, Roche is investigating the efficacy and safety of Vabysmo in people with macular edema following retinal vein occlusion in two phase III studies, BALATON and COMINO.29,30 Roche has also initiated several phase IV studies, including the Elevatum study of Vabysmo in underrepresented patient populations with DME, the SALWEEN study of Vabysmo in a subpopulation of nAMD highly prevalent in Asia, as well as the VOYAGER study, a global real-world data collection platform.31-33 Roche also supports several other independent studies to further understand retinal conditions with a high unmet need
Vabysmo is the first bispecific antibody approved for the eye.34,35 It targets and inhibits two signalling pathways linked to a number of vision-threatening retinal conditions by neutralising angiopoietin-2 (Ang-2) and vascular endothelial growth factor-A (VEGF-A).36,37 Ang-2 and VEGF-A contribute to vision loss by destabilising blood vessels, causing new leaky blood vessels to form and increasing inflammation.36,37 By blocking pathways involving Ang-2 and VEGF-A, Vabysmo is designed to stabilise blood vessels. Vabysmo is approved in 60 countries around the world, including the United States (U.S.), Japan, the United Kingdom and in the European Union for people living with neovascular or ‘wet’ age-related macular degeneration and diabetic macular edema. Review by other regulatory authorities is ongoing.
Roche is focused on saving people’s eyesight from the leading causes of vision loss through pioneering therapies. Through our innovation in the scientific discovery of new potential drug targets, personalised healthcare, molecular engineering, biomarkers and continuous drug delivery, we strive to design the right therapies for the right patients.
We have the broadest retina pipeline in ophthalmology, which is led by science and informed by insights from people with eye diseases. Our pipeline includes gene therapies and treatments for geographic atrophy and other vision-threatening diseases, including rare and inherited conditions.
Applying our extensive experience, we have already brought breakthrough ophthalmic treatments to people living with vision loss. Susvimo™ (ranibizumab injection) 100 mg/mL for intravitreal use via ocular implant is the first U.S. Food and Drug Administration-approved refillable eye implant for neovascular or ‘wet’ age-related macular degeneration that continuously delivers a customised formulation of ranibizumab over a period of months.40 Vabysmo® (faricimab) is the first bispecific antibody approved for the eye, which targets two signalling pathways that drive retinal conditions.34,37 Lucentis®* (ranibizumab injection) is the first treatment approved to improve vision in people with certain retinal conditions.
Roche is a global pioneer in pharmaceuticals and diagnostics focused on advancing science to improve people’s lives. The combined strengths of pharmaceuticals and diagnostics under one roof have made Roche the leader in personalised healthcare – a strategy that aims to fit the right treatment to each patient in the best way possible.
Roche is the world’s largest biotech company, with truly differentiated medicines in oncology, immunology, infectious diseases, ophthalmology and diseases of the central nervous system. Roche is also the world leader in in vitro diagnostics and tissue-based cancer diagnostics, and a frontrunner in diabetes management.
Founded in 1896, Roche continues to search for better ways to prevent, diagnose and treat diseases and make a sustainable contribution to society. The company also aims to improve patient access to medical innovations by working with all relevant stakeholders. More than thirty medicines developed by Roche are included in the World Health Organization Model Lists of Essential Medicines, among them life-saving antibiotics, antimalarials and cancer medicines. Moreover, for the twelfth consecutive year, Roche has been recognised as one of the most sustainable companies in the Pharmaceuticals Industry by the Dow Jones Sustainability Indices (DJSI).
The Roche Group, headquartered in Basel, Switzerland, is active in over 100 countries and in 2020 employed more than 100,000 people worldwide. In 2020, Roche invested CHF 12.2 billion in R&D and posted sales of CHF 58.3 billion. Genentech, in the United States, is a wholly owned member of the Roche Group. Roche is the majority shareholder in Chugai Pharmaceutical, Japan
IMbrave050 is a Phase III global, multicentre, open-label, randomised study evaluating the efficacy and safety of adjuvant Tecentriq plus Avastin, compared with active surveillance, in people with HCC at high risk of recurrence (determined by the size and number of cancerous lesions and the histopathology results, if available) after surgical resection or ablation with curative intent.
The study randomised 668 people with a ratio of 1:1 to receive either Tecentriq (1,200 mg every three weeks) plus Avastin (15 mg/kg every three weeks) for a period of 12 months or 17 cycles, or no intervention with active surveillance. The primary endpoint is independent review facility-assessed RFS. Key secondary endpoints include OS, RFS as determined by the investigator and RFS in patients with PD-L1-positive disease.
Liver cancer is the third leading cause of cancer death and one of the few cancers where mortality is rising.4,5 More than 900,000 people are diagnosed with the disease globally each year, which translates to one person diagnosed every 90 seconds.4 Nine out of ten cases of HCC are caused by chronic liver disease, which includes chronic hepatitis B and C infection, non-alcoholic fatty liver disease (NAFLD), non-alcoholic steatohepatitis (NASH), alcohol-related liver disease (ALD) and cirrhosis resulting from these conditions.5,6
If diagnosed in the early stage, surgery may be prescribed to remove the primary tumour, however an estimated 70-80% of people with early-stage HCC experience disease recurrence following surgery.2 Early recurrence is associated with poorer prognosis and shorter survival.2,7 Tumour size, number of tumours, and portal vein invasion are associated with an increased risk of recurrence.
Tecentriq is a cancer immunotherapy approved for some of the most aggressive and difficult-to-treat forms of cancer. Tecentriq was the first cancer immunotherapy approved for the treatment of a certain type of early-stage non-small cell lung cancer (NSCLC), small cell lung cancer (SCLC) and HCC. Tecentriq is also approved in countries around the world, either alone or in combination with targeted therapies and/or chemotherapies, for various forms of metastatic NSCLC, certain types of metastatic urothelial cancer, PD-L1-positive metastatic triple-negative breast cancer and BRAF V600 mutation-positive advanced melanoma.
Tecentriq is a monoclonal antibody designed to bind with a protein called programmed death ligand-1 (PD-L1), which is expressed on tumour cells and tumour-infiltrating immune cells, blocking its interactions with both PD-1 and B7.1 receptors. By inhibiting PD-L1, Tecentriq may enable the activation of T-cells. Tecentriq is a cancer immunotherapy that has the potential to be used as a foundational combination partner with other immunotherapies, targeted medicines and various chemotherapies across a broad range of cancers. In addition to intravenous infusion, the formulation of Tecentriq is also being investigated as a subcutaneous injection to help address the growing burden of cancer treatment for patients and healthcare systems.
Roche is a global pioneer in pharmaceuticals and diagnostics focused on advancing science to improve people’s lives. The combined strengths of pharmaceuticals and diagnostics under one roof have made Roche the leader in personalised healthcare – a strategy that aims to fit the right treatment to each patient in the best way possible.
Roche is the world’s largest biotech company, with truly differentiated medicines in oncology, immunology, infectious diseases, ophthalmology and diseases of the central nervous system. Roche is also the world leader in in vitro diagnostics and tissue-based cancer diagnostics, and a frontrunner in diabetes management.
Founded in 1896, Roche continues to search for better ways to prevent, diagnose and treat diseases and make a sustainable contribution to society. The company also aims to improve patient access to medical innovations by working with all relevant stakeholders. More than thirty medicines developed by Roche are included in the World Health Organization Model Lists of Essential Medicines, among them life-saving antibiotics, antimalarials and cancer medicines. Moreover, for the twelfth consecutive year, Roche has been recognised as one of the most sustainable companies in the Pharmaceuticals Industry by the Dow Jones Sustainability Indices (DJSI).
The Roche Group, headquartered in Basel, Switzerland, is active in over 100 countries and in 2020 employed more than 100,000 people worldwide. In 2020, Roche invested CHF 12.2 billion in R&D and posted sales of CHF 58.3 billion. Genentech, in the United States, is a wholly owned member of the Roche Group. Roche is the majority shareholder in Chugai Pharmaceutical, Japan