In the YOSEMITE and RHINE studies in diabetic macular edema, at least 60% of eligible Vabysmo patients could extend treatment to every four months at two years, compared to 50% at year one
Almost 80% of eligible Vabysmo patients could extend treatment to every three months or longer in both studies
In the Archway study in neovascular age-related macular degeneration, 95% of Susvimo patients maintained a six-month treatment schedule at two years
Basel, 11 February 2022 - Roche (SIX: RO, ROG; OTCQX: RHHBY) today announced that new two-year data from its phase III studies of Vabysmo™ (faricimab) and Susvimo™ (previously called Port Delivery System with ranibizumab) will be presented at Angiogenesis, Exudation and Degeneration 2022 on 12 February. These longer-term results from the Vabysmo YOSEMITE and RHINE studies in diabetic macular edema (DME) and the Susvimo Archway study in neovascular or “wet” age-related macular degeneration (nAMD) further reinforce the potential to allow for longer time between treatments and fewer eye injections for people with these conditions, while still achieving and maintaining vision gains seen with previous standard-of-care injections. Neovascular AMD and DME are two leading causes of vision loss, together affecting around 40 million people worldwide, which require treatment with eye injections as often as once a month.1-4
“Results from these three studies reinforce the potential of Vabysmo and Susvimo to redefine standards of care and reduce treatment burden for people living with diabetic macular edema and neovascular AMD,” said Levi Garraway, M.D., Ph.D., Roche’s Chief Medical Officer and Head of Global Product Development. “These two first-of-their-kind treatments are the culmination of over a decade of pioneering research, aiming to better address the needs of people with retinal conditions.”
In the YOSEMITE and RHINE studies, at least 60% of people eligible for extended dosing with Vabysmo could be treated every four months at two years – a 10 percentage point increase since the primary analyses at one year – while achieving non-inferior vision gains versus aflibercept given every two months. Furthermore, nearly 80% of people eligible for extended dosing with Vabysmo could be treated every three months or longer. In the Archway study, Susvimo allowed 95% of people to go six months between treatments at two years – the fourth complete refill-exchange interval – while maintaining vision outcomes that were non-inferior to monthly ranibizumab injections. Across all three studies, with longer follow-up, Vabysmo and Susvimo continued to be generally well-tolerated, with favourable benefit-risk profiles. Safety will continue to be monitored closely in the post-market setting.
VabysmoTM (faricimab-svoa) is the first bispecific antibody for the eye approved by the U.S. Food and Drug Administration (FDA), and the only injectable eye medicine approved for treatments from one to four months apart in the first year following four initial monthly doses, based on evaluation of the patient’s anatomy and vision outcomes.5 Vabysmo is designed to block two disease pathways linked to a number of vision-threatening retinal conditions by neutralising angiopoietin-2 (Ang-2) and vascular endothelial growth factor-A (VEGF-A). Ang-2 and VEGF-A are thought to contribute to vision loss by destabilising blood vessels, which may cause new leaky blood vessels to form and increase inflammation. While additional research continues, inhibition of both pathways has been shown in preclinical studies to have potentially complementary benefits, stabilising vessels and thereby reducing vessel leakage and inflammation more than inhibition of VEGF-A alone.4
Susvimo is the first nAMD treatment in 15 years to provide an alternative to standard-of-care eye injections. By continuously delivering medicine into the eye through a refillable implant, SusvimoTM (ranibizumab injection) 100 mg/mL for intravitreal use via ocular implant is the only FDA-approved treatment that may help people with nAMD maintain their vision with as few as two treatments per year
In the YOSEMITE and RHINE studies, DME patients received Vabysmo, given either every two months or up to every four months using a treat and extend approach, or aflibercept given every two months. Two-year results showed Vabysmo patients maintained the vision improvements achieved in the first year and vision gains continued to be non-inferior to those achieved by aflibercept patients. In YOSEMITE, the average vision gains from baseline at two years were +10.7 eye chart letters in both the Vabysmo treat and extend and two-month arms, and +11.4 letters in the aflibercept arm. In RHINE, the average vision gains from baseline at two years were +10.1 and +10.9 letters in the Vabysmo treat and extend and two-month arms, respectively, and +9.4 letters in the aflibercept arm.
Importantly, 60% (n=162/270) of Vabysmo treat and extend patients in YOSEMITE and 64.5% (n=185/287) in RHINE achieved four-month dosing at two years. This is an increase over one-year results, which showed 52.8% (n=151/286) of Vabysmo treat and extend patients in YOSEMITE and 51% (n=157/308) in RHINE achieved four-month dosing. An additional 18.1% (n=49/270) of Vabysmo treat and extend patients in YOSEMITE and 13.6% (n=39/287) in RHINE achieved three-month dosing. Combined, almost 80% of Vabysmo treat and extend patients were able to go three months or longer between treatments at the end of the second year. Across study arms, Vabysmo showed consistent two-step or better improvement in diabetic retinopathy according to the Early Treatment Diabetic Retinopathy Study – Diabetic Retinopathy Severity Score (ETDRS-DRSS). At two years, 42.8% of Vabysmo treat and extend patients in YOSEMITE and 44.3% in RHINE achieved a two-step or better improvement from baseline. In the two-month Vabysmo arms, 51.4% and 53.5% of patients in YOSEMITE and RHINE, respectively, achieved a two-step or better improvement in diabetic retinopathy severity. Vabysmo given at intervals of up to four months continued to demonstrate greater reductions in central subfield thickness (CST) compared to aflibercept given every two months in both studies. Safety results were consistent across study arms, with no reported cases of retinal vasculitis or retinal occlusive events.
One-year results from the YOSEMITE and RHINE studies and the TENAYA and LUCERNE studies in nAMD were recently published in The Lancet.
Neovascular AMD patients in Archway received either Susvimo refilled every six months or monthly ranibizumab 0.5 mg eye injections. Two-year results showed vision was maintained by Susvimo patients and continued to be non-inferior to that achieved with monthly ranibizumab injections. Susvimo patients averaged -1.1 eye chart letters in visual acuity from baseline at two years, while monthly ranibizumab patients averaged -0.5 letters from baseline. In addition, 95% of Susvimo patients were able to go six months without needing additional treatment in the second, third and fourth refill-exchange intervals. In Archway, Susvimo was generally well-tolerated, with a favourable benefit-risk profile. The most common adverse events of special interest (≥5%) were cataract, conjunctival bleb and vitreous haemorrhage. The safety profile of Susvimo in the clinical trial setting is well understood and will continue to be monitored closely.
In addition to Archway results, two-year interim data from the ongoing phase III Portal study will be presented at the Angiogenesis meeting. Portal is an extension study evaluating the long-term safety and efficacy of Susvimo in nAMD.
Vabysmo is approved by the FDA for the treatment of nAMD and DME.5 Susvimo is approved by the FDA for the treatment of people with nAMD who have previously responded to at least two anti-VEGF injections.6 Vabysmo is currently under review by the European Medicines Agency for the treatment of nAMD and DME and Susvimo is under review for the treatment of nAMD. Submissions to other regulatory authorities around the world are ongoing.
Roche has a robust phase III clinical development programme for Vabysmo and Susvimo. For Vabysmo, the programme includes AVONELLE-X, an extension study of TENAYA and LUCERNE evaluating the long-term safety and tolerability of Vabysmo in nAMD, and RHONE-X, an extension study of YOSEMITE and RHINE evaluating the long-term safety and tolerability of Vabysmo in DME.8,9 Additionally, the COMINO and BALATON trials are also underway, evaluating the efficacy and safety of Vabysmo in people with macular edema following retinal vein occlusion.10,11
For Susvimo, the clinical development programme includes the Portal, Pagoda, Pavilion and Velodrome studies.12-15 Portal is an extension study evaluating the long-term safety and efficacy of Susvimo in nAMD.12 Pagoda is evaluating Susvimo for the treatment of DME, while Pavilion is a study of Susvimo in diabetic retinopathy without DME.13,14 Velodrome is evaluating Susvimo refilled every nine months in nAMD
YOSEMITE (NCT03622580) and RHINE (NCT03622593) are two identical, randomised, multicentre, double-masked, global phase III studies evaluating the efficacy and safety of Vabysmo™ (faricimab) compared to aflibercept in 1,891 people with diabetic macular edema (940 in YOSEMITE and 951 in RHINE). The studies each have three treatment arms: Vabysmo 6.0 mg administered up to every four months after four initial monthly doses using a treat and extend approach; Vabysmo 6.0 mg administered at two-month intervals after six initial monthly doses; and aflibercept administered at fixed two-month intervals after five initial monthly doses. Dosing schedule for patients within the treat-and-extend arm was determined by central subfield thickness (CST) and visual acuity. In all three arms, sham injections were administered at study visits when treatment injections were not scheduled to maintain the masking of investigators and participants.
The primary endpoint of the studies is the average change in best-corrected visual acuity (BCVA) score (the best distance vision a person can achieve – including with correction such as glasses – when reading letters on an eye chart) from baseline at one year, averaged over weeks 48, 52 and 56. Secondary endpoints include: safety; the percentage of participants in the treat and extend arm receiving Vabysmo every one, two, three and four months, at week 52; the percentage of participants achieving a two-step or greater improvement from baseline in diabetic retinopathy severity at week 52; the percentage of participants achieving a gain, and the percentage avoiding a loss, of 15 letters or more in BCVA from baseline over time; change in CST from baseline over time; and percentage of patients with absence of intraretinal fluid over time.
Archway (NCT03677934) was a randomised, multicentre, open-label phase III study evaluating the efficacy and safety of Susvimo™ (previously called Port Delivery System with ranibizumab), refilled every six months at fixed intervals, compared to monthly intravitreal injections of ranibizumab 0.5 mg in 415 people living with neovascular or “wet” age-related macular degeneration. Patients enrolled in Archway were responders to prior treatment with anti-vascular endothelial growth factor (VEGF) therapy. In both study arms, patients were treated with at least three anti-VEGF injections within the six months prior to their Archway screening visit. The primary endpoint of the study was the change in best-corrected visual acuity (BCVA) score from baseline at the average of Week 36 and Week 40. Secondary endpoints include safety, overall change in vision (BCVA) from baseline and change from baseline in centre point thickness over time.
Age-related macular degeneration (AMD) is a condition that affects the part of the eye that provides sharp, central vision needed for activities like reading.1 Neovascular or “wet” AMD (nAMD) is an advanced form of the disease that can cause rapid and severe vision loss.17,18 It develops when new and abnormal blood vessels grow uncontrolled under the macula, causing swelling, bleeding and/or fibrosis.18 Worldwide, around 20 million people are living with nAMD – the leading cause of vision loss in people over the age of 60 – and the condition will affect even more people around the world as the global population ages.
Affecting around 21 million people globally, diabetic macular edema (DME) is a vision-threatening condition associated with blindness and decreased quality of life when left untreated.3,20 DME occurs when the damaged blood vessels leak into and cause swelling in the macula – the central area of the retina responsible for the sharp vision needed for reading and driving.21,22 The number of people with DME is expected to grow as the prevalence of diabetes increases.23 There remains a significant unmet need for more effective, longer-lasting therapies for people with DME.
Vabysmo™ (faricimab) is the first bispecific antibody designed for the eye. It targets and inhibits two disease pathways linked to a number of vision-threatening retinal conditions by neutralising angiopoietin-2 (Ang-2) and vascular endothelial growth factor-A (VEGF-A). Ang-2 and VEGF-A contribute to vision loss by destabilising blood vessels, causing new leaky blood vessels to form and increasing inflammation. By blocking both pathways involving Ang-2 and VEGF-A, Vabysmo is designed to stabilise blood vessels.
Susvimo™ (previously called Port Delivery System with ranibizumab) is a refillable eye implant surgically inserted into the eye during a one-time, outpatient procedure. Susvimo continuously delivers a customised formulation of ranibizumab over time. Susvimo is indicated for intravitreal use via the Susvimo eye implant only. Ranibizumab is a vascular endothelial growth factor (VEGF) inhibitor designed to bind to and inhibit VEGF-A, a protein that has been shown to play a critical role in the formation of new blood vessels and the leakiness of the vessels.6
Susvimo is different from the ranibizumab intravitreal injection, a medicine marketed as Lucentis®* (ranibizumab injection), which is approved to treat nAMD and other retinal diseases. Lucentis* was first approved for nAMD by the FDA in 2006
Roche is focused on saving people’s eyesight from the leading causes of vision loss through pioneering therapies. Through our innovation in the scientific discovery of new potential drug targets, personalised healthcare, molecular engineering, biomarkers and continuous drug delivery, we strive to design the right therapies for the right patients.
We have the broadest retina pipeline in Ophthalmology, which is led by science and informed by insights from people with eye diseases. Our pipeline includes gene therapies and treatments for geographic atrophy and other vision-threatening diseases, including rare and inherited conditions.
Applying our extensive experience, we have already brought breakthrough ophthalmic treatments to people living with vision loss. Susvimo™ (ranibizumab injection) 100 mg/mL for intravitreal use via ocular implant is the first U.S. Food and Drug Administration (FDA)-approved refillable eye implant for neovascular or “wet” age-related macular degeneration that continuously delivers a customised formulation of ranibizumab over a period of months.6 Vabysmo™ (faricimab-svoa) is the first FDA-approved bispecific antibody for the eye, which targets two distinct pathways that drive retinal conditions.5 Lucentis®️* (ranibizumab injection) is the first treatment approved to improve vision in people with certain retinal conditions.
Roche is a global pioneer in pharmaceuticals and diagnostics focused on advancing science to improve people’s lives. The combined strengths of pharmaceuticals and diagnostics under one roof have made Roche the leader in personalised healthcare – a strategy that aims to fit the right treatment to each patient in the best way possible.
Roche is the world’s largest biotech company, with truly differentiated medicines in oncology, immunology, infectious diseases, ophthalmology and diseases of the central nervous system. Roche is also the world leader in in vitro diagnostics and tissue-based cancer diagnostics, and a frontrunner in diabetes management.
Founded in 1896, Roche continues to search for better ways to prevent, diagnose and treat diseases and make a sustainable contribution to society. The company also aims to improve patient access to medical innovations by working with all relevant stakeholders. More than thirty medicines developed by Roche are included in the World Health Organization Model Lists of Essential Medicines, among them life-saving antibiotics, antimalarials and cancer medicines. Moreover, for the twelfth consecutive year, Roche has been recognised as one of the most sustainable companies in the Pharmaceuticals Industry by the Dow Jones Sustainability Indices (DJSI).
The Roche Group, headquartered in Basel, Switzerland, is active in over 100 countries and in 2020 employed more than 100,000 people worldwide. In 2020, Roche invested CHF 12.2 billion in R&D and posted sales of CHF 58.3 billion. Genentech, in the United States, is a wholly owned member of the Roche Group. Roche is the majority shareholder in Chugai Pharmaceutical, Japan
Vabysmo (faricimab-svoa) targets and inhibits two disease pathways that drive neovascular or “wet” age-related macular degeneration (nAMD) and diabetic macular edema (DME)
Vabysmo is the only injectable eye medicine approved simultaneously in the US for nAMD and DME, with flexible dosing regimens based on patient need
Basel, 31 January 2022 - Roche (SIX: RO, ROG; OTCQX: RHHBY) today announced that the U.S. Food and Drug Administration (FDA) has approved Vabysmo™ (faricimab-svoa) for the treatment of neovascular or “wet” age-related macular degeneration (nAMD) and diabetic macular edema (DME). Neovascular AMD and DME are two leading causes of vision loss worldwide.1 Vabysmo targets and inhibits two disease pathways linked to a number of vision-threatening retinal conditions by neutralising angiopoietin-2 (Ang-2) and vascular endothelial growth factor-A (VEGF-A).2 Vabysmo is the first and only FDA-approved injectable eye medicine for nAMD and DME that improves and maintains vision with treatments from one to four months apart in the first year following four initial monthly doses, based on evaluation of the patient’s anatomy and vision outcomes.3 Standard of care for nAMD and DME typically requires eye injections every one to two months.2,4
“Vabysmo represents an important step forward for ophthalmology. It is the first bispecific antibody approved for the eye and a major advance in treating retinal conditions such as neovascular AMD and diabetic macular edema,” said Charles Wykoff, M.D., Ph.D., Director of Research at Retina Consultants of Texas in Houston and a Vabysmo phase III investigator. “With Vabysmo, we now have the opportunity to offer patients a medicine that could improve their vision, potentially lowering treatment burden with fewer injections over time.”
The approval is based on positive results across four phase III studies in nAMD and DME. The studies consistently showed that patients treated with Vabysmo given at intervals of up to four months achieved non-inferior vision gains versus aflibercept given every two months in the first year. Vabysmo was generally well tolerated in all four studies, with a favourable benefit-risk profile.2,4 The most common adverse reaction (≥5%) reported in patients receiving Vabysmo was conjunctival hemorrhage (7%).3 Two scientific papers and an editorial on these one-year results were recently published in The Lancet.2,4
Vabysmo is designed to block pathways involving Ang-2 and VEGF-A. Ang-2 and VEGF-A are thought to contribute to vision loss by destabilising blood vessels, which may cause new leaky blood vessels to form and increase inflammation. While additional research continues, inhibition of both pathways has been shown in preclinical studies to have potentially complementary benefits, stabilising vessels, and thereby reducing vessel leakage and inflammation.2
“Vabysmo provides a new approach to treating vision-threatening retinal conditions through a mechanism of action that targets two pathways simultaneously,” said Levi Garraway, M.D., Ph.D., Roche’s Chief Medical Officer and Head of Global Product Development. “This is our second FDA approval in ophthalmology in recent months, underscoring our commitment to people living with retinal conditions.”
With Vabysmo, people with nAMD initially receive four monthly treatments. Based on anatomical and vision outcomes, they may receive subsequent treatments every two, three or four months. People with DME are initially given four monthly treatments. Subsequently, their treatment may be extended or reduced based on anatomical and vision outcomes, with a range of one to four months between doses. A second approved treatment regimen for DME involves six monthly loading doses, followed by treatment every two months. Some people with nAMD and DME may be treated monthly if needed, although additional efficacy was not demonstrated in most people given Vabysmo every month.3
Roche has ongoing long-term extension studies for Vabysmo in people with nAMD and DME. These include AVONELLE-X, an extension study of TENAYA and LUCERNE evaluating the long-term safety and tolerability of Vabysmo in nAMD, and RHONE-X, an extension study of YOSEMITE and RHINE evaluating the long-term safety and tolerability of Vabysmo in DME.5,6 Additionally, the COMINO and BALATON trials are also underway, evaluating the efficacy and safety of Vabysmo in people with macular edema following retinal vein occlusion.7,8
Vabysmo will be available in the United States in the coming weeks. The European Medicines Agency is also currently evaluating the Vabysmo Marketing Authorisation Application for the treatment of nAMD and DME.
TENAYA (NCT03823287) and LUCERNE (NCT03823300) are two identical, randomised, multicentre, double-masked, global phase III studies evaluating the efficacy and safety of Vabysmo compared to aflibercept in 1,329 people living with neovascular or “wet” age-related macular degeneration (671 in TENAYA and 658 in LUCERNE). The studies each have two treatment arms: Vabysmo 6.0 mg administered at intervals of two, three, or four months, following four initial monthly doses, selected based on objective assessment of disease activity at weeks 20 and 24; and aflibercept 2.0 mg administered at fixed two-month intervals after three initial monthly doses. In both arms, sham injections were administered at study visits when treatment injections were not scheduled, to maintain the masking of investigators and participants. The primary endpoint of the studies is the average change in best-corrected visual acuity (BCVA) score (the best distance vision a person can achieve – including with correction such as glasses – when reading letters on an eye chart) from baseline averaged over weeks 40, 44 and 48. Secondary endpoints include: safety; the percentage of participants in the Vabysmo arm receiving treatment every two, three and four months; the percentage of participants achieving a gain, and the percentage avoiding a loss, of 15 letters or more in BCVA from baseline over time; change in central subfield thickness (CST) from baseline over time; and change in total area of choroidal neovascularisation (CNV) lesion and leakage from baseline over time.
Both studies met their primary endpoint, with Vabysmo given at intervals of up to every four months consistently shown to offer visual acuity gains that were non-inferior to aflibercept given every two months. In TENAYA and LUCERNE, the average vision gains from baseline at one year in the Vabysmo arms were +5.8 and +6.6 letters, respectively, compared to +5.1 and +6.6 letters in the aflibercept arms.
The studies also measured the proportion of people in the Vabysmo arm that were treated on dosing schedules of every three or four months during the first year. In both studies, comparable reductions in CST and CNV size and area of leakage were observed with Vabysmo given at intervals of up to four months versus aflibercept given every two months in the first year. Vabysmo was generally well-tolerated in both studies, with a favourable benefit-risk profile. In TENAYA and LUCERNE, the most common adverse reactions (≥3% of patients) included conjunctival haemorrhage, vitreous floaters, retinal pigment epithelial tears, increase of intraocular pressure and eye pain.3 Safety results were consistent across study arms.
YOSEMITE (NCT03622580) and RHINE (NCT03622593) are two identical, randomised, multicentre, double-masked, global phase III studies evaluating the efficacy and safety of Vabysmo compared to aflibercept in 1,891 people with diabetic macular edema (940 in YOSEMITE and 951 in RHINE). The studies each have three treatment arms: Vabysmo 6.0 mg administered up to every four months after four initial monthly doses using a treat-and-extend approach; Vabysmo 6.0 mg administered at two-month intervals after six initial monthly doses; and aflibercept administered at fixed two-month intervals after five initial monthly doses. In all three arms, sham injections were administered at study visits when treatment injections were not scheduled, to maintain the masking of investigators and participants.
The primary endpoint of the studies is the average change in BCVA score from baseline at one year, averaged over weeks 48, 52 and 56. Secondary endpoints included: safety; the percentage of participants in the treat-and-extend arm receiving Vabysmo every one, two, three and four months at week 52; the percentage of participants achieving a two-step or greater improvement from baseline in diabetic retinopathy severity at week 52; the percentage of participants achieving a gain, and the percentage avoiding a loss, of 15 letters or more in BCVA from baseline over time; change in central subfield thickness (CST) from baseline over time; and percentage of patients with absence of intraretinal fluid over time.
Both studies met their primary endpoint, with Vabysmo given at intervals of up to every four months consistently shown to offer visual acuity gains that were non-inferior to aflibercept given every two months. In YOSEMITE, the average vision gains from baseline at one year were +11.6 and +10.7 eye chart letters in the Vabysmo treat-and-extend and two-month arms, respectively, and +10.9 letters in the aflibercept arm. In RHINE, the average vision gains from baseline at one year were +10.8 and +11.8 letters in the Vabysmo treat-and-extend and two-month arms, respectively, and +10.3 letters in the aflibercept arm.
A secondary endpoint in both studies measured the proportion of people in the Vabysmo treat-and-extend arm that achieved dosing schedules of every three or four months at the end of the first year. In both studies, greater reductions in CST and intraretinal fluid were observed with Vabysmo given at intervals of up to four months versus aflibercept given every two months in the first year. Vabysmo was generally well-tolerated in both studies, with a favourable benefit-risk profile. In YOSEMITE and RHINE, the most common adverse reactions (≥3% of patients) included conjunctival haemorrhage, vitreous floaters and increase of intraocular pressure.3 Safety results were consistent across study arms.
Age-related macular degeneration (AMD) is a condition that affects the part of the eye that provides sharp, central vision needed for activities like reading.9,10 Neovascular or “wet” AMD (nAMD) is an advanced form of the disease that can cause rapid and severe vision loss if left untreated. It develops when new and abnormal blood vessels grow uncontrolled under the macula, causing swelling, bleeding and/or fibrosis. Worldwide, around 20 million people are living with nAMD – the leading cause of vision loss in people over the age of 60 – and the condition will affect even more people around the world as the global population ages
Affecting around 21 million people globally, diabetic macular edema (DME) is a vision-threatening retinal condition associated with blindness and decreased quality of life when left untreated.15,16 DME occurs when damaged blood vessels in the retina leak into and cause swelling in the macula – the central area of the retina responsible for the sharp vision needed for reading and driving.10,17 The number of people with DME is expected to grow as the prevalence of diabetes increases.18 There remains a significant unmet need for more effective, longer-lasting therapies for people with DME
Vabysmo (faricimab-svoa) is the first bispecific antibody approved for the eye. It targets and inhibits two disease pathways linked to a number of vision-threatening retinal conditions by neutralising angiopoietin-2 (Ang-2) and vascular endothelial growth factor-A (VEGF-A). Ang-2 and VEGF-A contribute to vision loss by destabilising blood vessels, causing new leaky blood vessels to form and increasing inflammation. By blocking pathways involving Ang-2 and VEGF-A, Vabysmo is designed to stabilise blood vessels.
Roche is focused on saving people’s eyesight from the leading causes of vision loss through pioneering therapies. Through our innovation in the scientific discovery of new potential drug targets, personalised healthcare, molecular engineering, biomarkers, and continuous drug delivery, we strive to design the right therapies for the right patients.
We have the broadest retina pipeline in ophthalmology, which is led by science and informed by insights from people with eye diseases. Our pipeline includes gene therapies and treatments for geographic atrophy and other vision-threatening diseases, including rare and inherited conditions.
Applying our extensive experience, we have already brought breakthrough ophthalmic treatments to people living with vision loss. Susvimo™ (ranibizumab injection) 100 mg/mL for intravitreal use via ocular implant is the first FDA-approved refillable eye implant for neovascular or “wet” age-related macular degeneration that continuously delivers a customised formulation of ranibizumab over a period of months.19 Vabysmo™ (faricimab-svoa), the first FDA-approved bispecific antibody for the eye, which targets two disease pathways that drive retinal conditions.3 Lucentis®️* (ranibizumab injection) is the first treatment approved to improve vision in people with certain retinal conditions
Roche is a global pioneer in pharmaceuticals and diagnostics focused on advancing science to improve people’s lives. The combined strengths of pharmaceuticals and diagnostics under one roof have made Roche the leader in personalised healthcare – a strategy that aims to fit the right treatment to each patient in the best way possible.
Roche is the world’s largest biotech company, with truly differentiated medicines in oncology, immunology, infectious diseases, ophthalmology and diseases of the central nervous system. Roche is also the world leader in in vitro diagnostics and tissue-based cancer diagnostics, and a frontrunner in diabetes management.
Founded in 1896, Roche continues to search for better ways to prevent, diagnose and treat diseases and make a sustainable contribution to society. The company also aims to improve patient access to medical innovations by working with all relevant stakeholders. More than thirty medicines developed by Roche are included in the World Health Organization Model Lists of Essential Medicines, among them life-saving antibiotics, antimalarials and cancer medicines. Moreover, for the twelfth consecutive year, Roche has been recognised as one of the most sustainable companies in the Pharmaceuticals Industry by the Dow Jones Sustainability Indices (DJSI).
The Roche Group, headquartered in Basel, Switzerland, is active in over 100 countries and in 2020 employed more than 100,000 people worldwide. In 2020, Roche invested CHF 12.2 billion in R&D and posted sales of CHF 58.3 billion. Genentech, in the United States, is a wholly owned member of the Roche Group. Roche is the majority shareholder in Chugai Pharmaceutical, Japan
Interim data submitted to the FDA show majority of pre-symptomatic babies treated with Evrysdi for at least one year were able to sit, stand and walk within timeframes typical of healthy babies, as well as maintain swallowing
Evrysdi is approved in 70 countries and submitted in a further 31 with more than 4,500 patients treated to date
Basel, 25 January 2022 – Roche (SIX: RO, ROG; OTCQX: RHHBY) today announced that the U.S. Food and Drug Administration (FDA) has granted priority review of a supplemental new drug application (sNDA) for the use of Evrysdi® (risdiplam) to treat pre-symptomatic babies under two months of age with spinal muscular atrophy (SMA). The sNDA submission incorporates interim data from the RAINBOWFISH study, which shows the majority of pre-symptomatic babies treated with Evrysdi achieved key milestones such as sitting, standing, walking and maintained the ability to swallow following 12 months of treatment.
“Treating very young babies with Evrysdi before SMA symptoms arise may help them to achieve milestones such as standing and walking within timeframes typical of healthy infants,” said Levi Garraway, M.D., Ph. D., Roche’s Chief Medical Officer and Head of Global Product Development. “Extending treatment access for the youngest members of the SMA community is crucial and we look forward to working with the FDA on this application.”
Evrysdi is designed to treat SMA by increasing and sustaining production of the survival motor neuron (SMN) protein in the central nervous system (CNS) and peripheral tissues. SMN protein is found throughout the body and is critical for maintaining healthy motor neurons and movement. Evrysdi’s existing FDA label is for the treatment of SMA in adults, children and babies two months and older. If approved, Evrysdi would be the first medicine administered at-home for pre-symptomatic babies with SMA.
Initial interim data from the RAINBOWFISH study, presented at the World Muscle Society (WMS) Virtual Congress 2021, showed that of the babies included in the interim efficacy analysis, all (5/5), maintained the ability to swallow and were able to feed exclusively orally after 12 months of treatment. Eighty per cent (4/5) treated with Evrysdi for at least 12 months achieved milestones such as standing and walking independently within World Health Organization windows for healthy children. All participants (n=5) met HINE-2* motor milestones of head control, sitting upright, rolling and crawling after 12 months of treatment with Evrysdi.
No treatment related serious adverse events were reported in any of the babies treated with Evrysdi through the interim safety analysis period (n=12). Four treatment emergent adverse events were reported, and all were resolved or were resolving with ongoing treatment with Evrysdi. The most common adverse events (AEs) were nasal congestion (33%), cough (25%), teething (25%), vomiting (25%), eczema (17%), abdominal pain (17%), diarrhoea (17%), gastroenteritis (17%), papule (17%) and pyrexia (17%). The AEs were reflective of the age of the babies rather than the underlying SMA. The RAINBOWFISH study is currently recruiting.
The latest results from the RAINBOWFISH study will be presented at the Muscular Dystrophy Association (MDA) Clinical and Scientific Conference in March 2022.
Roche leads the clinical development of Evrysdi as part of a collaboration with the SMA Foundation and PTC Therapeutics.
Evrysdi is a survival motor neuron 2 (SMN2) splicing modifier designed to treat SMA caused by mutations in chromosome 5q that lead to SMN protein deficiency. Evrysdi is administered daily at home in liquid form by mouth or by feeding tube.
Evrysdi is designed to treat SMA by increasing and sustaining the production of the survival motor neuron (SMN) protein in the central nervous system (CNS) and peripheral tissues. SMN protein is found throughout the body and is critical for maintaining healthy motor neurons and movement.
Evrysdi was granted PRIME designation by the European Medicines Agency (EMA) in 2018 and Orphan Drug Designation by the U.S Food and Drug Administration in 2017. In 2021 Evrysdi was awarded Drug Discovery of the Year by the British Pharmacological Society as well as the Society for Medicines Research award for Drug Discovery. Evrysdi is currently approved in 70 countries and the dossier is under review in a further 31 countries.
Evrysdi is currently being evaluated in five multicentre trials in people with SMA:
FIREFISH (NCT02913482) – an open-label, two-part pivotal clinical trial in infants with Type 1 SMA. Part 1 was a dose-escalation study in 21 infants with the primary objective of assessing the safety profile of risdiplam in infants and determining the dose for Part 2. Part 2 is a pivotal, single-arm study of risdiplam in 41 infants with Type 1 SMA treated for 2 years, followed by an open-label extension. Enrolment for Part 2 was completed in November 2018. The primary objective of Part 2 was to assess efficacy as measured by the proportion of infants sitting without support after 12 months of treatment, as assessed by the Gross Motor Scale of the Bayley Scales of Infant and Toddler Development – Third Edition (BSID-III) (defined as sitting without support for 5 seconds). The study met its primary endpoint.
SUNFISH (NCT02908685) – SUNFISH is a two part, double-blind, placebo controlled pivotal study in people aged 2-25 years with Types 2 or 3 SMA. Part 1 (n=51) determined the dose for the confirmatory Part 2. Part 2 (n=180) evaluated motor function using the total score of Motor Function Measure 32 (MFM-32) at 12 months. MFM-32 is a validated scale used to evaluate fine and gross motor function in people with neurological disorders, including SMA. The study met its primary endpoint.
JEWELFISH (NCT03032172) – an open-label exploratory trial designed to assess the safety, tolerability, pharmacokinetics and pharmacodynamics in people with SMA aged 6 months to 60 years (inclusion criteria) who received other investigational or approved SMA therapies for at least 90 days prior to receiving Evrysdi. The study has completed recruitment (n=174).
RAINBOWFISH (NCT03779334) – an open-label, single-arm, multicentre study, investigating the efficacy, safety, pharmacokinetics and pharmacodynamics of risdiplam in babies (~n=25), from birth to six weeks of age (at first dose) with genetically diagnosed SMA who are not yet presenting with symptoms. The study is currently recruiting.
MANATEE (NCT05115110) – a global phase 2/3 clinical study to evaluate the safety and efficacy of GYM329 (RO7204239), an anti-myostatin molecule targeting muscle growth, in combination with Evrysdi for the treatment of SMA in patients 2-10 years of age. The FDA Office of Orphan Products Development granted GYM329 Orphan Drug Designation for the treatment of patients with SMA in December 2021. The study is commencing recruitment in Q1 2022.
SMA is a severe, progressive neuromuscular disease that can be fatal. It affects approximately one in 10,000 babies and is the leading genetic cause of infant mortality. SMA is caused by a mutation of the survival motor neuron 1 (SMN1) gene, which leads to a deficiency of SMN protein. This protein is found throughout the body and is essential to the function of nerves that control muscles and movement. Without it, nerve cells cannot function correctly, leading to muscle weakness over time. Depending on the type of SMA, an individual’s physical strength and their ability to walk, eat or breathe can be significantly diminished or lost.
Neuroscience is a major focus of research and development at Roche. Our goal is to pursue groundbreaking science to develop new treatments that help improve the lives of people with chronic and potentially devastating diseases.
Roche is investigating more than a dozen medicines for neurological disorders, including multiple sclerosis, neuromyelitis optica spectrum disorder, Alzheimer’s disease, Huntington’s disease, Parkinson’s disease, Duchenne muscular dystrophy and autism spectrum disorder. Together with our partners, we are committed to pushing the boundaries of scientific understanding to solve some of the most difficult challenges in neuroscience today.
Roche is a global pioneer in pharmaceuticals and diagnostics focused on advancing science to improve people’s lives. The combined strengths of pharmaceuticals and diagnostics under one roof have made Roche the leader in personalised healthcare – a strategy that aims to fit the right treatment to each patient in the best way possible.
Roche is the world’s largest biotech company, with truly differentiated medicines in oncology, immunology, infectious diseases, ophthalmology and diseases of the central nervous system. Roche is also the world leader in in vitro diagnostics and tissue-based cancer diagnostics, and a frontrunner in diabetes management.
Founded in 1896, Roche continues to search for better ways to prevent, diagnose and treat diseases and make a sustainable contribution to society. The company also aims to improve patient access to medical innovations by working with all relevant stakeholders. More than thirty medicines developed by Roche are included in the World Health Organization Model Lists of Essential Medicines, among them life-saving antibiotics, antimalarials and cancer medicines. Moreover, for the twelfth consecutive year, Roche has been recognised as one of the most sustainable companies in the Pharmaceuticals Industry by the Dow Jones Sustainability Indices (DJSI).
The Roche Group, headquartered in Basel, Switzerland, is active in over 100 countries and in 2020 employed more than 100,000 people worldwide. In 2020, Roche invested CHF 12.2 billion in R&D and posted sales of CHF 58.3 billion. Genentech, in the United States, is a wholly owned member of the Roche Group. Roche is the majority shareholder in Chugai Pharmaceutical, Japan
Roche’s new cloud-based platform connects patient test results, medical records and third-party applications, allowing professionals to monitor patients’ health, adjust treatment protocols, and make quick data-driven patient care decisions anytime, anywhere within and across clinical settings.
The new digital ecosystem breaks down common information silos for point of care (POC) professionals, reducing costs and administrative burdens by connecting information securely and compliantly across devices, providers, and locations.
Cobas® infinity edge is designed as an open platform, allowing healthcare professionals to easily access and adopt third-party innovators’ new digital tools and medically-relevant applications via the Roche digital marketplace.
Basel, 11 January 2022 - Roche (SIX: RO, ROG; OTCQX: RHHBY) today introduced cobas® infinity edge, a secure and encrypted cloud-based platform for the integration and management of point of care data, available for use in clinical settings globally.
More people are using digital technologies in health than ever before. Only 15% of healthcare organisations are fully equipped to make quick data-driven decisions due to lack of data integration, multiple incompatible IT systems, and other information silos. With longer life expectancies, and more people living with chronic conditions, healthcare systems face growing pressure to manage more patient data and diagnostic information. Digital health ecosystems can help address these challenges.
The cobas® infinity edge platform supports healthcare professionals in overcoming these challenges by bringing multiple capabilities together in one digital health ecosystem. Using applications, it facilitates professionals’ ability to view and manage patient test results compliantly, while allowing the integration of this data into patients’ electronic medical records (EMRs). It allows deploying new applications in a point of care device, such as the cobas® pulse glucose meter, and connecting patient health information from this device in an easy, timely, secure way to various IT systems. Concurrently, it allows health professionals to access a marketplace of medically relevant applications to drive adoption of digital tools that improve patient care, by leveraging Roche’s network of third party developers. Bringing all of these capabilities together, it breaks down information silos, ultimately driving improved patient care and operational efficiency within and across point of care clinical settings and hospital departments.
“Roche is pioneering digital innovations to solve operational and clinical challenges in healthcare,” says Thomas Schinecker, CEO Roche Diagnostics. “With cobas® infinity edge, we are introducing the infrastructure for a digital ecosystem in point of care. We have combined our ability to connect health data safely, securely and compliantly plus our deep healthcare expertise with a unique marketplace, where professionals can use Roche or third-party digital health solutions. We can accelerate digital adoption in healthcare and deliver access to health information anytime, anywhere, for improving patient care.”
The cobas® infinity edge solution suite is compliant with ISO27001, the international standard for information security for cloud-based products and compliant with GDPR and HIPAA data privacy regulations. Its digital ecosystem includes technologies to solve specific operational challenges in lab and hospital settings as well as a digital marketplace offering health-relevant applications from Roche and third-party innovative developers.
Following first commercial availability in select markets, Roche Diagnostics plans to offer the new digital platform in other markets globally throughout 2022 and 2023.
The Roche software solution suite called cobas® infinity edge includes three modules: unite, scribe, and smart. Used together, the solution facilitates the ability of labs and hospitals to manage patient-testing devices (Roche and non-Roche) that measure clinical health parameters anytime, anywhere. cobas® infinity edge also enables the deployment of third party applications into point of care devices. With this digital ecosystem approach, lab and hospital administrators can also better govern, update and manage use of near-patient testing devices - even remotely. The platform uses industry standard security technologies and processes to connect to electronic medical record systems (EMRs) as well as enabling remote installations, service and maintenance of devices outside the lab/hospital premises (e.g., clinics or pharmacies). This solution will bring more diagnostic capabilities - safely and securely - beyond the lab walls - closer to physicians and nurses at the points where clinical decisions are being made.
With growing investments in digital capabilities, Roche is building on its expertise in science, medicine and technology to increasingly leverage data analytic tools and novel digital technologies to transform the way healthcare is delivered and managed for each patient. Our data-driven digital solutions empower labs, healthcare providers and patients to make informed, insights-based decisions across the entire care continuum – reducing costs to society and improving patient health outcomes in the process.
Roche is a global pioneer in pharmaceuticals and diagnostics focused on advancing science to improve people’s lives. The combined strengths of pharmaceuticals and diagnostics under one roof have made Roche the leader in personalised healthcare – a strategy that aims to fit the right treatment to each patient in the best way possible.
Roche is the world’s largest biotech company, with truly differentiated medicines in oncology, immunology, infectious diseases, ophthalmology and diseases of the central nervous system. Roche is also the world leader in in vitro diagnostics and tissue-based cancer diagnostics, and a frontrunner in diabetes management.
Founded in 1896, Roche continues to search for better ways to prevent, diagnose and treat diseases and make a sustainable contribution to society. The company also aims to improve patient access to medical innovations by working with all relevant stakeholders. More than thirty medicines developed by Roche are included in the World Health Organization Model Lists of Essential Medicines, among them life-saving antibiotics, antimalarials and cancer medicines. Moreover, for the twelfth consecutive year, Roche has been recognised as one of the most sustainable companies in the Pharmaceuticals Industry by the Dow Jones Sustainability Indices (DJSI).
The Roche Group, headquartered in Basel, Switzerland, is active in over 100 countries and in 2020 employed more than 100,000 people worldwide. In 2020, Roche invested CHF 12.2 billion in R&D and posted sales of CHF 58.3 billion. Genentech, in the United States, is a wholly owned member of the Roche Group. Roche is the majority shareholder in Chugai Pharmaceutical, Japan
Results to be presented for the first time show mosunetuzumab induces high and durable complete response rates in people with follicular lymphoma who have received two or more prior therapies
New efficacy data from Roche’s portfolio of cancer immunotherapies demonstrate potential of bispecific antibodies to expand upon current treatment options across several blood cancers
Basel, 11 December 2021 - Roche (SIX: RO, ROG; OTCQX: RHHBY) today announced that new pivotal data on its CD20xCD3 T-cell engaging bispecific antibody, mosunetuzumab, will be presented for the first time at the 63rd American Society of Hematology (ASH) Annual Meeting and Exposition from 11-14 December 2021.
Emerging data continue to show the promising benefit-risk profile of mosunetuzumab in relapsed or refractory (R/R) follicular lymphoma (FL), a slow-growing, or indolent, form of non-Hodgkin lymphoma (NHL). Pivotal results from the phase I/II GO29781 study demonstrated that mosunetuzumab induces durable complete responses lasting at least 18 months in heavily pretreated patients with R/R FL who have received two or more prior therapies, with a 60.0% complete response (CR) rate and a median progression-free survival of 17.9 months (95% CI: 10.1-not evaluable). Median duration of response was 22.8 months among responders (95% CI: 9.7-not evaluable). The most common adverse event (AE) was cytokine release syndrome (CRS), which was generally low grade (mainly Grade 1-2).1
“Despite initial successful treatment, many people with follicular lymphoma often experience relapse. Mosunetuzumab could potentially become a highly efficacious treatment option that can be administered without the need for cell collection or genetic engineering,” said Levi Garraway, M.D., Ph.D., Roche’s Chief Medical Officer and Head of Global Product Development. “With mosunetuzumab, we also aim to offer a therapy that can be administered in the outpatient setting to people with this devastating blood cancer.”
Roche recently submitted the initial marketing authorisation application for mosunetuzumab to the European Medicines Agency, with the hope to bring this drug as soon as possible to people with NHL. Genentech plans to submit the new data to the U.S. Food and Drug Administration in the near future for approval consideration. If approved, mosunetuzumab has the potential to be a first-in-class CD20xCD3 T-cell engaging bispecific antibody in NHL.
Additionally, as part of Roche’s broad pipeline of haematology immunotherapies and application of novel combinations, key data for the bispecific antibodies mosunetuzumab, glofitamab and cevostamab are being presented, including:
Initial results from the phase Ib CO41942 study of mosunetuzumab in combination with lenalidomide in people with R/R FL who have received at least one prior line of therapy demonstrated encouraging preliminary efficacy and a tolerable safety profile.
Data from the phase Ib/II GO40516 study evaluating mosunetuzumab in combination with Polivy® (polatuzumab vedotin) showed promising efficacy and favourable safety in heavily pretreated patients with aggressive R/R NHL with an objective response rate (ORR) of 65.0% and a CR rate of 48.3%. CRS occurred in 18% of patients, and all events occurred in Cycle 1 and were Grade 1-2.3 A phase I/Ib NP30179 dose-escalation study evaluating glofitamab as a monotherapy and in combination with Gazyva®/Gazyvaro® (obinutuzumab) following pretreatment with Gazyva/Gazyvaro in patients with R/R B-cell NHL showed promising activity in both R/R FL and R/R mantle cell lymphoma (MCL), an uncommon but aggressive form of lymphoma with poor prognosis for those who progress.
Preliminary results in heavily pretreated patients with R/R FL showed high response rates across all treatment groups, including high-risk subgroups, with an ORR of 81.0% for the glofitamab monotherapy group and an ORR of 100% for the glofitamab plus Gazyva/Gazyvaro combination therapy group.5 For patients with R/R MCL, treated with glofitamab monotherapy following Gazyva/Gazyvaro pretreatment, the ORR was 81.0%.6 Across both studies, the most common AE was CRS, with the majority of events being low grade (Grade 1-2).5,6
Results of the phase Ib/II NP39488 study of glofitamab in combination with Polivy demonstrated encouraging preliminary efficacy and a tolerable safety profile in people with difficult-to-treat R/R diffuse large B-cell lymphoma. With a median follow up of 3.2 months (95% CI: 1.4-3.5), an ORR of 73.0% was observed with a 51.5% CR rate, with patients showing durable responses at ≥6 months. No Grade 3 or higher CRS events were observed, and the safety profile of the combination was consistent with that of the individual medicines.
Data from the phase I GO39775 dose-escalation and expansion study investigating cevostamab in heavily pretreated patients with R/R multiple myeloma (MM) showed the first-of-its kind FcRH5xCD3 bispecific antibody induced clinically meaningful, target dose-dependent increases in ORR without an increase in the rate of CRS, with an ORR of 54.5% in the 160 mg dose group. Results from double step-up dosing suggest this approach could help mitigate CRS and potentially improve the safety profile compared to single step-up dosing.
Our investigational cancer immunotherapies, mosunetuzumab and glofitamab, are T-cell engaging bispecific antibodies designed to engage with CD3 on the T cell and CD20 on the tumour cell, bringing them close in proximity and enabling the T cell to eliminate the tumour cell. Although these bispecific antibodies have similar modes of action, they differ in their structure and clinical profiles. Cevostamab, another investigational T-cell engaging bispecific antibody, is designed to target FcRH5 on myeloma cells and CD3 on T cells and is currently being evaluated in people living with R/R MM.
Roche’s broad and comprehensive clinical development programme will continue to evaluate mosunetuzumab, glofitamab and cevostamab as monotherapies and in combination with other established and/or novel therapies for malignant haematological conditions with the goal of providing treatment solutions tailored to the patient journey for each disease.
Roche is currently developing two T-cell engaging bispecific antibodies, mosunetuzumab and glofitamab, designed to target CD20 on the surface of B cells and CD3 on the surface of T cells. This dual targeting activates and redirects a patient’s existing T cells to engage and eliminate target B cells by releasing cytotoxic proteins into the B cells. Mosunetuzumab and glofitamab differ in their structures, and both are being developed by Roche as part of our ongoing strategy to explore multiple bispecific formats in order to identify those that maximise potential clinical benefits for patients. Mosunetuzumab has a structure similar to that of a natural human antibody in that it has two ‘Fab’ regions but is different from naturally-occurring antibodies in that one ‘Fab’ region targets CD20 and the other ‘Fab’ region targets CD3. Glofitamab is based on a novel structural format that we call ‘2:1,’ which refers to the structure of the antibody. It is engineered to have two ‘Fab’ regions that bind to CD20 and one ‘Fab’ region that binds to CD3. The clinical development programmes for mosunetuzumab and glofitamab include ongoing investigations of these molecules as monotherapies and in combination with other medicines for the treatment of people with CD20-positive B cell (non-Hodgkin lymphomas), including diffuse large B-cell lymphoma and follicular lymphoma .
Cevostamab (BFCR4350A) is an FcRH5xCD3 T-cell engaging bispecific antibody designed to target FcRH5 on myeloma cells and CD3 on T cells. FcRH5 is a unique and differentiated target, expressed on nearly all myeloma cells. Cevostamab has a structure similar to that of a natural human antibody in that it has two ‘Fab’ regions, but is different from naturally-occurring antibodies in that one ‘Fab’ region targets FcRH5 and the other ‘Fab’ region targets CD3. This dual targeting activates and re-directs a patient’s existing T cells to engage and eliminate target FcRH5-expressing myeloma cells by releasing cytotoxic proteins into the myeloma cells.
The GO29781 study [NCT02500407] is a phase I/II, multicentre, open-label, dose-escalation study evaluating the safety and pharmacokinetics of mosunetuzumab in people with relapsed or refractory B-cell non-Hodgkin lymphoma. Outcome measures include complete response rate (best response) by independent review facility (primary endpoint), objective response rate, duration of response, progression-free survival, safety and tolerability (secondary endpoints).
Roche has been developing medicines for people with malignant and non-malignant blood diseases for over 20 years; our experience and knowledge in this therapeutic area runs deep. Today, we are investing more than ever in our effort to bring innovative treatment options to patients across a wide range of haematologic diseases. Our approved medicines include MabThera®/Rituxan® (rituximab), Gazyva®/Gazyvaro® (obinutuzumab), Polivy® (polatuzumab vedotin), Venclexta®/Venclyxto® (venetoclax) in collaboration with AbbVie, and Hemlibra® (emicizumab). Our pipeline of investigational haematology medicines includes T-cell engaging bispecific antibodies, glofitamab and mosunetuzumab, targeting both CD20 and CD3, and cevostamab, targeting both FcRH5 and CD3; Tecentriq® (atezolizumab), a monoclonal antibody designed to bind with PD-L1; and crovalimab, an anti-C5 antibody engineered to optimise complement inhibition. Our scientific expertise, combined with the breadth of our portfolio and pipeline, also provides a unique opportunity to develop combination regimens that aim to improve the lives of patients even further.
Roche is a global pioneer in pharmaceuticals and diagnostics focused on advancing science to improve people’s lives. The combined strengths of pharmaceuticals and diagnostics under one roof have made Roche the leader in personalised healthcare – a strategy that aims to fit the right treatment to each patient in the best way possible.
Roche is the world’s largest biotech company, with truly differentiated medicines in oncology, immunology, infectious diseases, ophthalmology and diseases of the central nervous system. Roche is also the world leader in in vitro diagnostics and tissue-based cancer diagnostics, and a frontrunner in diabetes management.
Founded in 1896, Roche continues to search for better ways to prevent, diagnose and treat diseases and make a sustainable contribution to society. The company also aims to improve patient access to medical innovations by working with all relevant stakeholders. More than thirty medicines developed by Roche are included in the World Health Organization Model Lists of Essential Medicines, among them life-saving antibiotics, antimalarials and cancer medicines. Moreover, for the twelfth consecutive year, Roche has been recognised as one of the most sustainable companies in the Pharmaceuticals Industry by the Dow Jones Sustainability Indices (DJSI).
The Roche Group, headquartered in Basel, Switzerland, is active in over 100 countries and in 2020 employed more than 100,000 people worldwide. In 2020, Roche invested CHF 12.2 billion in R&D and posted sales of CHF 58.3 billion. Genentech, in the United States, is a wholly owned member of the Roche Group. Roche is the majority shareholder in Chugai Pharmaceutical, Japan
EMA’s CHMP recommended the approval yesterday
Approval based on results from four phase III studies in over 5,500 patients.
Actemra/RoActemra has also been provisionally approved in Australia for the treatment of COVID-19.
Basel, 07 December 2021 - Roche (SIX: RO, ROG; OTCQX: RHHBY) today announced that the European Commission has extended the marketing authorisation for Actemra®/RoActemra® (tocilizumab) to include the treatment of COVID-19 in adults who are receiving systemic corticosteroids and require supplemental oxygen or mechanical ventilation.1 This decision comes just hours after the recommendation by the European Medicines Agency's (EMA) Committee for Medicinal Products for Human Use (CHMP), reflecting the urgent need for Actemra/RoActemra as a potential treatment option during the COVID-19 public health emergency.
“Actemra/RoActemra is the second Roche medicine to have received rapid European Commission approval in COVID-19 in recent weeks,” said Levi Garraway, M.D., Ph.D., Roche’s Chief Medical Officer and Head of Global Product Development. “The totality of evidence shows that Actemra/RoActemra can benefit those suffering with severe COVID-19. Together with vaccines, other treatments and testing, Actemra/RoActemra forms an important piece of the care puzzle as we confront new challenges of the pandemic in Europe and around the world.”
The decision from the European Commission follows an accelerated assessment by the EMA’s CHMP, which reviewed results from four studies of Actemra/RoActemra in over 5,500 patients with severe or critical COVID-19. These include the Roche-led phase III COVACTA, EMPACTA and REMDACTA trials, and the University of Oxford’s Randomised Evaluation of COVID-19 Therapy (RECOVERY) study, which was supported by Roche.
Outside of the European Union, Actemra/RoActemra has been provisionally approved in Australia, authorised for emergency use in the United States and Ghana, and recommended by the World Health Organization (WHO) for the treatment of COVID-19.2,3,4,5 Roche is working closely with regulatory bodies and other partners around the world on the next steps to bring this medicine to as many people as possible.
Following the recent emergence of the new SARS-CoV-2 variant of concern, Omicron (B.1.1.529), WHO has reported that interleukin 6 receptor blockers, such as Actemra/RoActemra, are expected to still be effective for managing patients with severe COVID-19.6
In these exceptional times, we stand together with society, governments, healthcare providers and all those working towards the common goal of overcoming the COVID-19 pandemic.
Roche has evaluated Actemra/RoActemra in COVID-19 in three phase III randomised studies: COVACTA, EMPACTA and REMDACTA.
COVACTA was a global, randomised, double-blind, placebo-controlled phase III study (COVACTA, NCT04320615), which evaluated the safety and efficacy of intravenous Actemra/RoActemra plus standard of care in adult patients hospitalised with severe COVID-19 pneumonia compared to placebo plus standard of care. The primary and secondary endpoints included clinical status, mortality, mechanical ventilation and intensive care unit (ICU) variables. Patients were followed for 60 days post-randomisation.
EMPACTA (Evaluating Minority Patients with Actemra) was a phase III, randomised, double-blind, placebo-controlled multicentre study (EMPACTA, NCT04372186) which evaluated the efficacy and safety of Actemra/RoActemra in the treatment of COVID-19 pneumonia among hospitalised patients that are often underrepresented in clinical trials. The primary endpoint was the cumulative proportion of participants dying or requiring mechanical ventilation by Day 28. Secondary endpoints included: time to clinical failure (defined as the time to death), mechanical ventilation, ICU admission, or withdrawal (whichever occured first); mortality rate by Day 28; and time to hospital discharge or “ready for discharge.”
REMDACTA was a two-armed global phase III, randomised, double-blind, multicentre study (REMDACTA, NCT04409262) to evaluate the efficacy and safety of Actemra/RoActemra plus Veklury® (remdesivir), versus placebo plus Veklury in hospitalised patients with severe COVID-19 pneumonia receiving standard of care. Veklury is an antiviral medicine that works to stop replication of SARS-CoV-2, the virus that causes COVID-19. The REMDACTA trial was conducted in collaboration with Gilead Sciences, Inc. The primary endpoint was improvement in time to hospital discharge by Day 28. Key secondary endpoints included likelihood of death, likelihood of progression to mechanical ventilation or death, and clinical status. Clinical status was measured by the 7-category ordinal scale, which tracks patients’ clinical status based on the need for intensive care and/or ventilator use, as well as supplemental oxygen requirements. Patients were followed for 60 days post-randomisation.
There have also been a number of clinical trials with an external third party as the sponsor exploring the efficacy and safety of Actemra/RoActemra for the treatment of patients hospitalised with COVID-19, including the University of Oxford’s RECOVERY study, which was supported by Roche. RECOVERY was a phase III, randomised trial (NCT04381936), which evaluated whether multiple potential treatments, including Actemra/RoActemra, prevent death in hospitalised adult patients with severe COVID-19.
Results of a prospective meta-analysis of almost 11,000 patients across 27 clinical trials, published by researchers from the World Health Organization in The Journal of the American Medical Association, found that treatment of hospitalised patients with severe or critical COVID-19 with IL-6 receptor blockers, including Actemra/RoActemra, was associated with improved mortality and reduced progression to invasive mechanical ventilation or death compared with usual care or placebo. The prospective meta-analysis included data on Actemra/RoActemra in COVID-19 from COVACTA, EMPACTA and REMDACTA, along with 16 additional third-party studies.
Actemra/RoActemra was the first approved anti-IL-6 receptor biologic and is available in both intravenous (IV) and subcutaneous (SC) formulations for the treatment of adult patients with moderate-to-severe active rheumatoid arthritis (RA). Actemra/RoActemra can be used alone or with methotrexate (MTX) in adult RA patients who are intolerant to, or have failed to respond to, other disease-modifying anti-rheumatic drugs (DMARDs). In Europe, RoActemra IV and SC are also approved for use in adult patients with severe, active and progressive RA who previously have not been treated with MTX. Actemra/RoActemra IV and SC are also approved globally for polyarticular juvenile idiopathic arthritis (pJIA) and systemic juvenile idiopathic arthritis (sJIA) in children two years of age and older. Actemra/RoActemra SC is approved globally for giant cell arteritis (GCA), and Actemra/RoActemra IV is approved for the treatment of chimeric antigen receptor (CAR) T-cell-induced severe or life-threatening cytokine release syndrome (CRS) in people two years of age and older. Actemra/RoActemra was the first approved treatment for sJIA, GCA and CRS. Actemra SC is now approved in the United States for slowing the rate of decline in pulmonary function in adult patients with systemic sclerosis-associated interstitial lung disease (SSc-ILD). In addition to the above-mentioned indications, in Japan Actemra IV is also approved for the treatment of Castleman’s disease and adult Still's disease, and the Actemra SC formulation is approved for Takayasu arteritis. RoActemra is also approved in Europe for the treatment of COVID-19 in adults who are receiving systemic corticosteroids and require supplemental oxygen or mechanical ventilation.
Actemra/RoActemra is part of a co-development agreement with Chugai Pharmaceutical Co., Ltd and has been approved in Japan since April 2005. Actemra/RoActemra is approved in more than 110 countries worldwide.
As a leading healthcare company, we are doing all we can to support countries in their fight against COVID-19 and minimising its impact. We have developed a growing number of diagnostic solutions that help to detect and diagnose the infection, as well as providing digital support to healthcare systems. We also continue to identify, develop, and support therapies which can play a role in treating the disease.
The impact of COVID-19 goes beyond those who contract it. That is why we are working with healthcare providers, laboratories, authorities, and organisations to help make sure patients continue to receive the tests, treatment and care they need during these challenging times. Building on a longstanding tradition of partnerships, we are working together with governments and others to make healthcare stronger and more sustainable in the future.
Reliable, high-quality testing is essential to help healthcare systems overcome this pandemic and Roche has so far launched 21 diagnostics solutions to help minimise the impact of COVID-19. As soon as the novel SARS-CoV-2 virus was sequenced in early 2020, we got to work. On 13 March 2020 we became the first company to receive United States (U.S.) Food and Drug Administration (FDA) Emergency Use Authorization (EUA) for a high-volume molecular test to detect the virus. Since then, we have continued to add a range of diagnostics solutions to our global portfolio to help in the fight against COVID-19. In addition to the gold standard PCR test, we have developed antigen tests to help diagnose the virus in settings where there is limited molecular laboratory infrastructure, rapid antigen tests where the virus can be detected on the spot, tests that can test for both flu and COVID-19 at the same time, both high throughput and at the point of care, and tests that can detect virus antibodies that can help monitor the spread of the virus and can also support in vaccine development. In March 2021 the SARS-CoV-2 variant test was launched, designed to detect key spike mutations.
Aside from these tests we have also looked at how we can support care for patients who have COVID-19, receiving an U.S. FDA EUA for the Elecsys® IL-6 test to assist in identifying severe inflammatory response in patients with confirmed COVID-19, as well as launching Roche v-TAC, a digital algorithm that could help simplify the screening, diagnosis, and monitoring of respiratory-compromised patients with COVID-19. Roche is working closely with governments and health authorities around the world and has significantly increased production to support availability of tests globally.
Roche is also actively involved in understanding the potential of the existing pharmaceuticals portfolio and is researching options for the future. In 2020, Roche entered into a number of new partnerships, including with Regeneron and Gilead to develop, manufacture and distribute molecules that can potentially both treat and prevent COVID-19.
Roche entered a partnership with Regeneron to jointly develop Ronapreve™ (casirivimab and imdevimab, known as REGEN-COV™ in the US. The antibody combination has been approved for use in the European Union and Japan, and conditionally in the United Kingdom and Australia, and is authorised for emergency or temporary pandemic use in additional territories such as the US and Canada. In addition, the World Health Organization recommended the use of Ronapreve for the treatment of patients with COVID-19.
In June 2021, Actemra/RoActemra received an EUA from the U.S. FDA for the intravenous treatment of COVID-19 in hospitalised adults and paediatric patients (2 years of age and older) who are receiving systemic corticosteroids and require supplemental oxygen, non-invasive or invasive mechanical ventilation, or extracorporeal membrane oxygenation. In addition, the World Health Organization recommended the use of Actemra/RoActemra for the treatment of certain patients with COVID-19.
Roche is a global pioneer in pharmaceuticals and diagnostics focused on advancing science to improve people’s lives. The combined strengths of pharmaceuticals and diagnostics under one roof have made Roche the leader in personalised healthcare – a strategy that aims to fit the right treatment to each patient in the best way possible.
Roche is the world’s largest biotech company, with truly differentiated medicines in oncology, immunology, infectious diseases, ophthalmology and diseases of the central nervous system. Roche is also the world leader in in vitro diagnostics and tissue-based cancer diagnostics, and a frontrunner in diabetes management.
Founded in 1896, Roche continues to search for better ways to prevent, diagnose and treat diseases and make a sustainable contribution to society. The company also aims to improve patient access to medical innovations by working with all relevant stakeholders. More than thirty medicines developed by Roche are included in the World Health Organization Model Lists of Essential Medicines, among them life-saving antibiotics, antimalarials and cancer medicines. Moreover, for the twelfth consecutive year, Roche has been recognised as one of the most sustainable companies in the Pharmaceuticals Industry by the Dow Jones Sustainability Indices (DJSI).
The Roche Group, headquartered in Basel, Switzerland, is active in over 100 countries and in 2020 employed more than 100,000 people worldwide. In 2020, Roche invested CHF 12.2 billion in R&D and posted sales of CHF 58.3 billion. Genentech, in the United States, is a wholly owned member of the Roche Group. Roche is the majority shareholder in Chugai Pharmaceutical, Japan
The combination rapid antigen test quickly differentiates between SARS-CoV-2 and influenza viruses A and B infections, with results ready in less than 30 minutes, allowing informed decisions on patient and pandemic management decisions.
Affordable and small, instrument-free testing kit enables convenient use for healthcare professionals at different point of care locations and in resource-limited settings.
The test works seamlessly with NAVIFY® Pass, Roche’s digital solution that allows individuals and healthcare professionals to immediately store, display, and share their COVID-19 test results and vaccine status through a unique data matrix
Basel, 6 December 2021 - Roche (SIX: RO, ROG; OTCQX: RHHBY) today announced its plans to launch the SARS-CoV-2 & Flu A/B Rapid Antigen Test for professional use in markets accepting the CE Mark by the beginning of January. Roche also intends to file for Emergency Use Authorization (EUA) to the U.S. Food and Drug Administration (FDA) in early 2022.
The SARS-CoV-2 & Flu A/B Rapid Antigen Test is intended for use by healthcare professionals to rapidly differentiate between SARS-CoV-2 and influenza viruses A and B infections in individuals with symptoms consistent with COVID-19 or influenza. The single integrated combination test uses a nasopharyngeal swab specimen to produce qualitative results (“yes/no” answer) on the presence of SARS-CoV-2 and/or influenza A and/or B antigens in 15-30 minutes. The test has a relative sensitivity of 84.85% and specificity of 98.59% for SARS-CoV-2. For the flu, the test has a positive percent agreement of 81.16% (influenza virus A) and 100% (influenza virus B) against a molecular test, and relative specificity of 100% (influenza virus A) and 99.04% (influenza virus B).1 The currently available sequences of the Omicron variant SARS-CoV-2 (B.1.1.529) have been analyzed, and based on the initial in silico investigations, an impact on the performance of the test is not expected.
Equipping healthcare professionals with a single integrated combination test is beneficial in settings where timely clinical decisions are needed or where central laboratory testing is difficult to access. The SARS-CoV-2 & Flu A/B Rapid Antigen Test helps healthcare professionals at the point of care to quickly diagnose and differentiate infections with any of the three respiratory viruses and aid in pursuing appropriate courses of action, including patient and pandemic management decisions.
Thomas Schinecker, CEO of Roche Diagnostics, stated, “It is critical that healthcare professionals have the ability to quickly know whether a patient has an infection with either SARS-CoV-2 or the flu, especially as the COVID-19 pandemic extends into our flu season. The combination rapid antigen test will help ensure the right decisions are taken by healthcare providers to treat patients and ultimately prevent community spread. The test adds a solution that will be critical to healthcare systems’ long-term management of SARS-CoV-2 and seasonal flu, as we transition from today’s global health emergency to the endemic phase of tomorrow.”
Together with the SARS-CoV-2 & Flu A/B Rapid Antigen Test, Roche is offering NAVIFY® Pass. This digital solution allows individuals and healthcare professionals to remotely store, display, and share their COVID-19 test results and vaccine status. With a unique and personalised data matrix placed on the test, NAVIFY® Pass can automatically read out all details about the test and establish a connection between patients and their individual test results.
The launch will be in partnership with SD Biosensor Inc., with whom Roche has a global distribution agreement and previously launched the SARS-CoV-2 Rapid Antigen Tests (Nasopharyngeal/Nasal), SARS-CoV-2 Antigen Self Test Nasal, and SARS-CoV-2 Rapid Antibody Test in countries accepting the CE Mark throughout 2020 and 2021. The test will become the fifth rapid test and twenty-second addition to Roche’s comprehensive portfolio of diagnostic solutions to help healthcare systems across the globe combat the COVID-19 pandemic through laboratory testing and at the point of care. Roche Diagnostics’ portfolio includes a wide range of molecular, serological and digital solutions that help diagnose and manage COVID-19 during the initial stages of infections, during the recovery phase, and following the resolution of infection.
The SARS-CoV-2 & Flu A/B Rapid Antigen Test is a rapid chromatographic immunoassay for the simultaneous detection and differentiation of the nucleocapsid protein antigens of SARS-CoV-2, influenza virus A, and influenza virus B in human nasopharyngeal swab samples. This test is intended as an aid in the differential diagnosis of SARS-CoV-2, influenza virus A, and influenza virus B infections, in individuals suspected of respiratory viral infections consistent with COVID-19 or influenza, by healthcare providers within the first five days since the onset of symptoms.
The test has a relative specificity of 98.59% for SARS-CoV-2, 100% for influenza viruses A and 99.04% for influenza viruses B. The relative sensitivity for SARS-CoV-2 was 84.85%. These performance characteristics are based on a prospective study of 104 participants where two nasopharyngeal swab samples were collected per participant and results of the rapid antigen test were compared with those from a highly-sensitive, FDA-cleared RT-PCR method. The sensitivity for influenza viruses A and B was evaluated separately using banked human nasopharyngeal swab samples, due to the limited circulation of influenza viruses in the past season. The rapid antigen test results were compared to highly sensitive RT-PCR method results, and the positive percent agreement for influenza viruses A and B were 81.16% and 100%, respectively. A prospective clinical study is ongoing to evaluate the influenza performance using fresh clinical samples. This test is not for self-testing. It is intended for professional use in laboratory and near-patient testing environments.
An antigen test detects proteins which are structural or functional components of a pathogen and are very specific to that pathogen. In this case, the test would provide a qualitative “yes/no” answer on the presence of the antigen in the patient sample and can be offered as a rapid strip test that is performed by healthcare professionals at the point of care. If the target antigen (in this case the nucleocapsid protein) is present in sufficient concentrations in the sample, it will bind to specific antibodies and generate a visually detectable signal on the test strip, typically with results ready in 15 to 30 minutes. A rapid antigen test can reliably detect individuals with a high viral load allowing healthcare professionals to quickly identify those patients at the greatest risk of spreading the infection.
As a leading healthcare company, we are doing all we can to support countries in their fight against COVID-19 and minimising its impact. We have developed a growing number of diagnostic solutions that help to detect and diagnose the infection, as well as providing digital support to healthcare systems. We also continue to identify, develop, and support therapies which can play a role in treating the disease.
The impact of COVID-19 goes beyond those who contract it. That is why we are working with healthcare providers, laboratories, authorities, and organisations to help make sure patients continue to receive the tests, treatment and care they need during these challenging times. Building on a longstanding tradition of partnerships, we are working together with governments and others to make healthcare stronger and more sustainable in the future.
Reliable, high-quality testing is essential to help healthcare systems overcome this pandemic and Roche has so far launched 21 diagnostics solutions to help minimise the impact of COVID-19. As soon as the novel SARS-CoV-2 virus was sequenced in early 2020, we got to work. On 13 March 2020 we became the first company to receive U.S. Food and Drug Administration (FDA) Emergency Use Authorization (EUA) for a high-volume molecular test to detect the virus. Since then, we have continued to add a range of diagnostics solutions to our global portfolio to help in the fight against COVID-19. In addition to the gold standard PCR test, we have developed antigen tests to help diagnose the virus in settings where there is limited molecular laboratory infrastructure, rapid antigen tests where the virus can be detected on the spot, tests that can test for both flu and COVID-19 at the same time, both high throughput and at the point of care, and tests that can detect virus antibodies that can help monitor the spread of the virus and can also support in vaccine development. In March 2021 the SARS-CoV-2 variant test was launched, designed to detect key spike mutations.
Aside from these tests we have also looked at how we can support care for patients who have COVID-19, receiving an U.S. FDA EUA for the Elecsys® IL-6 test to assist in identifying severe inflammatory response in patients with confirmed COVID-19, as well as launching Roche v-TAC, a digital algorithm that could help simplify the screening, diagnosis, and monitoring of respiratory-compromised patients with COVID-19. Roche is working closely with governments and health authorities around the world, and has significantly increased production to support availability of tests globally.
Roche is also actively involved in understanding the potential of the existing pharmaceuticals portfolio and is researching options for the future. In 2020, Roche entered into a number of new partnerships, including with Regeneron and Gilead to develop, manufacture and distribute molecules that can potentially both treat and prevent COVID-19.
Roche entered a partnership with Regeneron to jointly develop Ronapreve™ (casirivimab and imdevimab, known as REGEN-COV™ in the United States [US]). The antibody combination has been approved for use in the European Union and Japan, and conditionally in the United Kingdom and Australia, and is authorised for emergency or temporary pandemic use in additional territories such as the US and Canada. In addition, the World Health Organization recommended the use of Ronapreve for the treatment of patients with COVID-19.
In addition, we have explored the potential of our existing medicine Actemra/RoActemra in three global phase III clinical trials investigating its safety and efficacy in COVID-19 associated pneumonia (COVACTA, EMPACTA and REMDACTA). In June 2021, Actemra/RoActemra received an EUA from the U.S. FDA for the intravenous treatment of COVID-19 in hospitalised adults and paediatric patients (2 years of age and older) who are receiving systemic corticosteroids and require supplemental oxygen, non-invasive or invasive mechanical ventilation, or extracorporeal membrane oxygenation. In addition, the World Health Organization recommended the use of Actemra/RoActemra for the treatment of certain patients with COVID-19.
SD Biosensor is a global in-vitro diagnostic company focused on the development of immunoassay and molecular diagnostic products at the POC. Founded in 2010, SD Biosensor has continued to research and develop products that can aid in the fast and accurate diagnosis of patients across the testing journey. Through these innovative products, they are striving to become a leading global in vitro diagnostics company.
Roche is a global pioneer in pharmaceuticals and diagnostics focused on advancing science to improve people’s lives. The combined strengths of pharmaceuticals and diagnostics under one roof have made Roche the leader in personalised healthcare – a strategy that aims to fit the right treatment to each patient in the best way possible.
Roche is the world’s largest biotech company, with truly differentiated medicines in oncology, immunology, infectious diseases, ophthalmology and diseases of the central nervous system. Roche is also the world leader in in vitro diagnostics and tissue-based cancer diagnostics, and a frontrunner in diabetes management.
Founded in 1896, Roche continues to search for better ways to prevent, diagnose and treat diseases and make a sustainable contribution to society. The company also aims to improve patient access to medical innovations by working with all relevant stakeholders. More than thirty medicines developed by Roche are included in the World Health Organization Model Lists of Essential Medicines, among them life-saving antibiotics, antimalarials and cancer medicines. Moreover, for the twelfth consecutive year, Roche has been recognised as one of the most sustainable companies in the Pharmaceuticals Industry by the Dow Jones Sustainability Indices (DJSI).
The Roche Group, headquartered in Basel, Switzerland, is active in over 100 countries and in 2020 employed more than 100,000 people worldwide. In 2020, Roche invested CHF 12.2 billion in R&D and posted sales of CHF 58.3 billion. Genentech, in the United States, is a wholly owned member of the Roche Group. Roche is the majority shareholder in Chugai Pharmaceutical, Japan
Actemra/RoActemra reduced the risk of death in patients with severe COVID-19, as evidenced by a review of four phase III studies
The European Commission is expected to make a final decision regarding approval in the near future
Basel, 06 December 2021 - Roche (SIX: RO, ROG; OTCQX: RHHBY) today announced that the European Medicines Agency’s (EMA) Committee for Medicinal Products for Human Use (CHMP) has recommended extending the marketing authorisation for Actemra®/RoActemra® (tocilizumab) to include the treatment of COVID-19 in adults who are receiving systemic corticosteroids and require supplemental oxygen or mechanical ventilation. A final decision regarding the approval of Actemra/RoActemra is expected from the European Commission in the near future.
“As COVID-19 cases in Europe rise and with pressure on hospitals likely to increase, the need for effective treatments for those suffering most severely with COVID-19 could intensify,” said Levi Garraway, M.D., Ph.D., Roche’s Chief Medical Officer and Head of Global Product Development. “We are proud that the CHMP has recognised the potential of Actemra/RoActemra as we continue our efforts to bring treatment options to those most in need.”
In August 2021, the EMA’s CHMP began an accelerated assessment of Actemra/RoActemra – this rapid analysis is reserved for medicines that may offer significant benefit to public health. The assessment reviewed results from four studies in over 5,500 patients with severe or critical COVID-19. These include the Roche-led phase III COVACTA, EMPACTA and REMDACTA trials, and the University of Oxford’s Randomised Evaluation of COVID-19 Therapy (RECOVERY) study, which was supported by Roche. The totality of this clinical evidence demonstrated that Actemra/RoActemra reduced the risk of death in patients with severe or critical COVID-19.1
Actemra/RoActemra has been provisionally approved in Australia, authorised for emergency use in the United States and Ghana, and recommended by the World Health Organization (WHO) for the treatment of COVID-19. 1,2,3,4 Roche is working closely with regulatory bodies and other partners around the world on the next steps to bring this medicine to as many people as possible.
Following the recent emergence of the new SARS-CoV-2 variant of concern, Omicron (B.1.1.529), WHO has reported that interleukin 6 receptor blockers, such as Actemra/RoActemra, are expected to still be effective for managing patients with severe COVID-19.5
In these exceptional times, Roche stands together with society, governments, healthcare providers and all those working towards the common goal of overcoming the COVID-19 pandemic.
Roche has evaluated Actemra/RoActemra in COVID-19 in three phase III randomised studies: COVACTA, EMPACTA and REMDACTA.
COVACTA was a global, randomised, double-blind, placebo-controlled phase III study (COVACTA, NCT04320615), which evaluated the safety and efficacy of intravenous Actemra/RoActemra plus standard of care in adult patients hospitalised with severe COVID-19 pneumonia compared to placebo plus standard of care. The primary and secondary endpoints included clinical status, mortality, mechanical ventilation and intensive care unit (ICU) variables. Patients were followed for 60 days post-randomisation.
EMPACTA (Evaluating Minority Patients with Actemra) was a phase III, randomised, double-blind, placebo-controlled multicentre study (EMPACTA, NCT04372186) which evaluated the efficacy and safety of Actemra/RoActemra in the treatment of COVID-19 pneumonia among hospitalised patients that are often underrepresented in clinical trials. The primary endpoint was the cumulative proportion of participants dying or requiring mechanical ventilation by Day 28. Secondary endpoints included: time to clinical failure (defined as the time to death), mechanical ventilation, ICU admission, or withdrawal (whichever occured first); mortality rate by Day 28; and time to hospital discharge or “ready for discharge.”
REMDACTA was a two-armed global phase III, randomised, double-blind, multicentre study (REMDACTA, NCT04409262) to evaluate the efficacy and safety of Actemra/RoActemra plus Veklury® (remdesivir), versus placebo plus Veklury in hospitalised patients with severe COVID-19 pneumonia receiving standard of care. Veklury is an antiviral medicine that works to stop replication of SARS-CoV-2, the virus that causes COVID-19. The REMDACTA trial was conducted in collaboration with Gilead Sciences, Inc. The primary endpoint was improvement in time to hospital discharge by Day 28. Key secondary endpoints included likelihood of death, likelihood of progression to mechanical ventilation or death, and clinical status. Clinical status was measured by the 7-category ordinal scale, which tracks patients’ clinical status based on the need for intensive care and/or ventilator use, as well as supplemental oxygen requirements. Patients were followed for 60 days post-randomisation.
There have also been a number of clinical trials with an external third party as the sponsor exploring the efficacy and safety of Actemra/RoActemra for the treatment of patients hospitalised with COVID-19, including the University of Oxford’s RECOVERY study, which was supported by Roche. RECOVERY was a phase III, randomised trial (NCT04381936), which evaluated whether multiple potential treatments, including Actemra/RoActemra, prevent death in hospitalised adult patients with severe COVID-19.
Results of a prospective meta-analysis of almost 11,000 patients across 27 clinical trials, published by researchers from the World Health Organization in The Journal of the American Medical Association, found that treatment of hospitalised patients with severe or critical COVID-19 with IL-6 receptor blockers, including Actemra/RoActemra, was associated with improved mortality and reduced progression to invasive mechanical ventilation or death compared with usual care or placebo. The prospective meta-analysis included data on Actemra/RoActemra in COVID-19 from COVACTA, EMPACTA and REMDACTA, along with 16 additional third-party studies.
Actemra/RoActemra was the first approved anti-IL-6 receptor biologic and is available in both intravenous (IV) and subcutaneous (SC) formulations for the treatment of adult patients with moderate-to-severe active rheumatoid arthritis (RA). Actemra/RoActemra can be used alone or with methotrexate (MTX) in adult RA patients who are intolerant to, or have failed to respond to, other disease-modifying anti-rheumatic drugs (DMARDs). In Europe, RoActemra IV and SC are also approved for use in adult patients with severe, active and progressive RA who previously have not been treated with MTX. Actemra/RoActemra IV and SC are also approved globally for polyarticular juvenile idiopathic arthritis (pJIA) and systemic juvenile idiopathic arthritis (sJIA) in children two years of age and older. Actemra/RoActemra SC is approved globally for giant cell arteritis (GCA), and Actemra/RoActemra IV is approved for the treatment of chimeric antigen receptor (CAR) T-cell-induced severe or life-threatening cytokine release syndrome (CRS) in people two years of age and older. Actemra/RoActemra was the first approved treatment for sJIA, GCA and CRS. Actemra SC is now approved in the United States for slowing the rate of decline in pulmonary function in adult patients with systemic sclerosis-associated interstitial lung disease (SSc-ILD). In addition to the above-mentioned indications, in Japan Actemra IV is also approved for the treatment of Castleman’s disease and adult Still's disease, and the Actemra SC formulation is approved for Takayasu arteritis.
Actemra/RoActemra is part of a co-development agreement with Chugai Pharmaceutical Co., Ltd and has been approved in Japan since April 2005. Actemra/RoActemra is approved in more than 110 countries worldwide.
As a leading healthcare company, we are doing all we can to support countries in their fight against COVID-19 and minimising its impact. We have developed a growing number of diagnostic solutions that help to detect and diagnose the infection, as well as providing digital support to healthcare systems. We also continue to identify, develop, and support therapies which can play a role in treating the disease.
The impact of COVID-19 goes beyond those who contract it. That is why we are working with healthcare providers, laboratories, authorities, and organisations to help make sure patients continue to receive the tests, treatment and care they need during these challenging times. Building on a longstanding tradition of partnerships, we are working together with governments and others to make healthcare stronger and more sustainable in the future.
Reliable, high-quality testing is essential to help healthcare systems overcome this pandemic and Roche has so far launched 21 diagnostics solutions to help minimise the impact of COVID-19. As soon as the novel SARS-CoV-2 virus was sequenced in early 2020, we got to work. On 13 March 2020 we became the first company to receive United States (U.S.) Food and Drug Administration (FDA) Emergency Use Authorization (EUA) for a high-volume molecular test to detect the virus. Since then, we have continued to add a range of diagnostics solutions to our global portfolio to help in the fight against COVID-19. In addition to the gold standard PCR test, we have developed antigen tests to help diagnose the virus in settings where there is limited molecular laboratory infrastructure, rapid antigen tests where the virus can be detected on the spot, tests that can test for both flu and COVID-19 at the same time, both high throughput and at the point of care, and tests that can detect virus antibodies that can help monitor the spread of the virus and can also support in vaccine development. In March 2021 the SARS-CoV-2 variant test was launched, designed to detect key spike mutations.
Aside from these tests we have also looked at how we can support care for patients who have COVID-19, receiving an U.S. FDA EUA for the Elecsys® IL-6 test to assist in identifying severe inflammatory response in patients with confirmed COVID-19, as well as launching Roche v-TAC, a digital algorithm that could help simplify the screening, diagnosis, and monitoring of respiratory-compromised patients with COVID-19. Roche is working closely with governments and health authorities around the world and has significantly increased production to support availability of tests globally.
Roche is also actively involved in understanding the potential of the existing pharmaceuticals portfolio and is researching options for the future. In 2020, Roche entered into a number of new partnerships, including with Regeneron and Gilead to develop, manufacture and distribute molecules that can potentially both treat and prevent COVID-19.
Roche entered a partnership with Regeneron to jointly develop Ronapreve™ (casirivimab and imdevimab, known as REGEN-COV™ in the US. The antibody combination has been approved for use in the European Union and Japan, and conditionally in the United Kingdom and Australia, and is authorised for emergency or temporary pandemic use in additional territories such as the US and Canada. In addition, the World Health Organization recommended the use of Ronapreve for the treatment of patients with COVID-19.
In June 2021, Actemra/RoActemra received an EUA from the U.S. FDA for the intravenous treatment of COVID-19 in hospitalised adults and paediatric patients (2 years of age and older) who are receiving systemic corticosteroids and require supplemental oxygen, non-invasive or invasive mechanical ventilation, or extracorporeal membrane oxygenation. In addition, the World Health Organization recommended the use of Actemra/RoActemra for the treatment of certain patients with COVID-19.
Roche is a global pioneer in pharmaceuticals and diagnostics focused on advancing science to improve people’s lives. The combined strengths of pharmaceuticals and diagnostics under one roof have made Roche the leader in personalised healthcare – a strategy that aims to fit the right treatment to each patient in the best way possible.
Roche is the world’s largest biotech company, with truly differentiated medicines in oncology, immunology, infectious diseases, ophthalmology and diseases of the central nervous system. Roche is also the world leader in in vitro diagnostics and tissue-based cancer diagnostics, and a frontrunner in diabetes management.
Founded in 1896, Roche continues to search for better ways to prevent, diagnose and treat diseases and make a sustainable contribution to society. The company also aims to improve patient access to medical innovations by working with all relevant stakeholders. More than thirty medicines developed by Roche are included in the World Health Organization Model Lists of Essential Medicines, among them life-saving antibiotics, antimalarials and cancer medicines. Moreover, for the twelfth consecutive year, Roche has been recognised as one of the most sustainable companies in the Pharmaceuticals Industry by the Dow Jones Sustainability Indices (DJSI).
The Roche Group, headquartered in Basel, Switzerland, is active in over 100 countries and in 2020 employed more than 100,000 people worldwide. In 2020, Roche invested CHF 12.2 billion in R&D and posted sales of CHF 58.3 billion. Genentech, in the United States, is a wholly owned member of the Roche Group. Roche is the majority shareholder in Chugai Pharmaceutical, Japan
Phase III POLARIX trial showed Polivy plus R-CHP was the first treatment in two decades to significantly improve outcomes in newly diagnosed diffuse large B-cell lymphoma (DLBCL) versus the standard of care.
Pivotal data on mosunetuzumab, a potential first-in-class CD20xCD3 T-cell engaging bispecific antibody, showed high response rates in relapsed or refractory follicular lymphoma (FL).
HAVEN 6 phase III interim data demonstrated Hemlibra’s favourable safety and efficacy profile in people with moderate or mild haemophilia A.
Basel, 23 November 2021 - Roche (SIX: RO, ROG; OTCQX: RHHBY) today announced that new data from its extensive haematology portfolio will be presented at the American Society of Hematology (ASH) Annual Meeting and Exposition from 11-14 December 2021. Roche molecules will be featured in more than 90 abstracts, including 17 oral presentations, showcasing new immunotherapies, unique treatment combinations, the application of novel endpoints, and fixed-duration regimens.
Results from three pivotal studies will be featured:
First presentation of efficacy and safety data from the phase III POLARIX study as a late-breaking abstract and in the ASH press programme. POLARIX met its primary endpoint of improving progression-free survival, showing Polivy® (polatuzumab vedotin) plus MabThera®/Rituxan® (rituximab), cyclophosphamide, doxorubicin, and prednisone (R-CHP) reduced the likelihood of disease worsening or death, versus the standard-of-care, MabThera/Rituxan plus cyclophosphamide, doxorubicin, vincristine and prednisone (R-CHOP), for people with newly diagnosed diffuse large B-cell lymphoma (DLBCL). The safety profile was comparable for Polivy plus R-CHP versus R-CHOP.1 POLARIX is being conducted in collaboration with The Lymphoma Study Association (LYSA) and The Lymphoma Academic Research Organisation (LYSARC).
Pivotal results from the phase I/II GO29781 study, presented for the first time and featured in the ASH press programme, showing mosunetuzumab, a CD20xCD3 T-cell engaging bispecific antibody immunotherapy, achieved high response rates with a manageable safety profile. These data suggest that it could be a new treatment option for people with relapsed or refractory follicular lymphoma (FL) who have received two or more prior therapies.2 FL is the most common indolent (slow growing) form of non-Hodgkin lymphoma, a type of blood cancer, which often returns after initial therapy.4
Interim data from the phase III HAVEN 6 study, which demonstrated the favourable safety and efficacy profile of Hemlibra® (emicizumab) in people with moderate or mild haemophilia A without factor VIII inhibitors.3 This patient population has historically not used prophylactic (preventative) treatments, likely due to delayed or missed diagnosis and a lack of treatments and treatment guidelines, meaning these patients have a significant unmet clinical need.5, 6
“For 20 years, we have remained committed to deepening our understanding of many benign and malignant blood disorders in order to better meet the urgent needs of patients with these diseases,” said Levi Garraway, M.D., Ph.D., Roche’s Chief Medical Officer and Head of Global Product Development. “Our data at ASH reinforce our conviction that following the science and developing versatile treatment approaches leads to improved outcomes for patients in increasingly meaningful ways.”
Data presented at ASH by Roche span numerous blood diseases, including lymphoma, leukaemia, multiple myeloma and haemophilia. Additional data to be presented include updated results for three T-cell engaging bispecific antibody immunotherapies: glofitamab and mosunetuzumab, targeting CD20 and CD3; and cevostamab, targeting FcRH5 and CD3.
Further information on the key abstracts featuring Roche medicines presented can be found in the table below.
Roche will be hosting a Live Media Event titled: ‘Redefining treatment standards to improve outcomes for patients with blood disorders’ on Wednesday 15 December, offering an exclusive opportunity to hear from experts about the pivotal data being presented. Register here to access the Roche Haematology Newsroom, available from Friday 26 November, where you’ll be able to attend the Live Media Event and find materials to further explain and provide context to Roche’s malignant and non-malignant data.
Follow Roche on Twitter via @Roche and LinkedIn and keep up to date with ASH Annual Meeting news and updates by using the hashtag #ASH21.
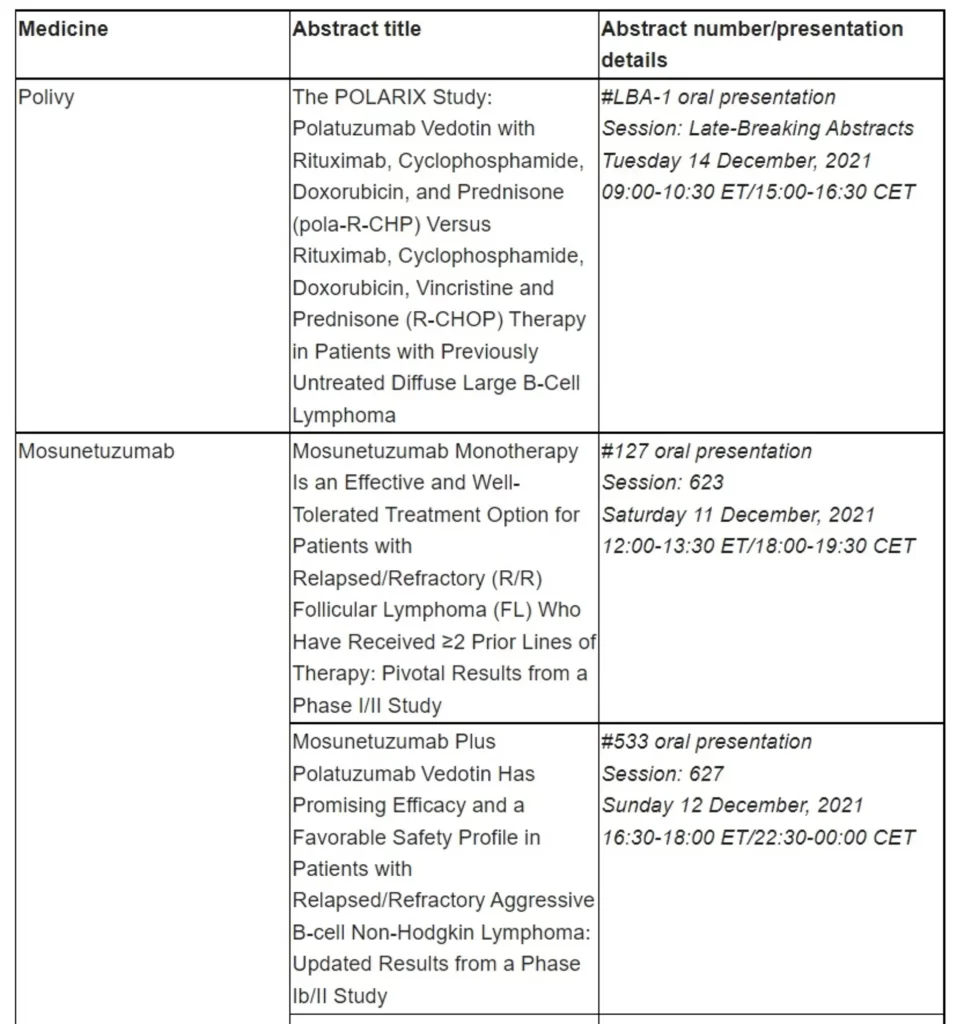
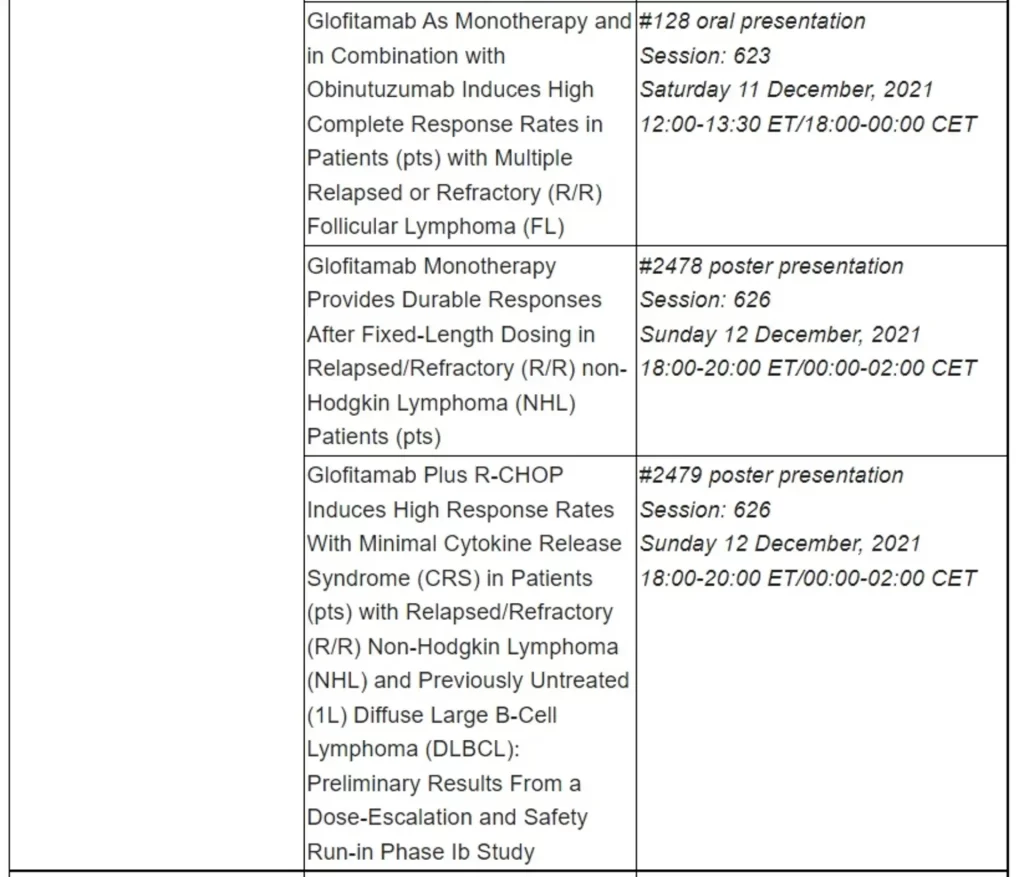
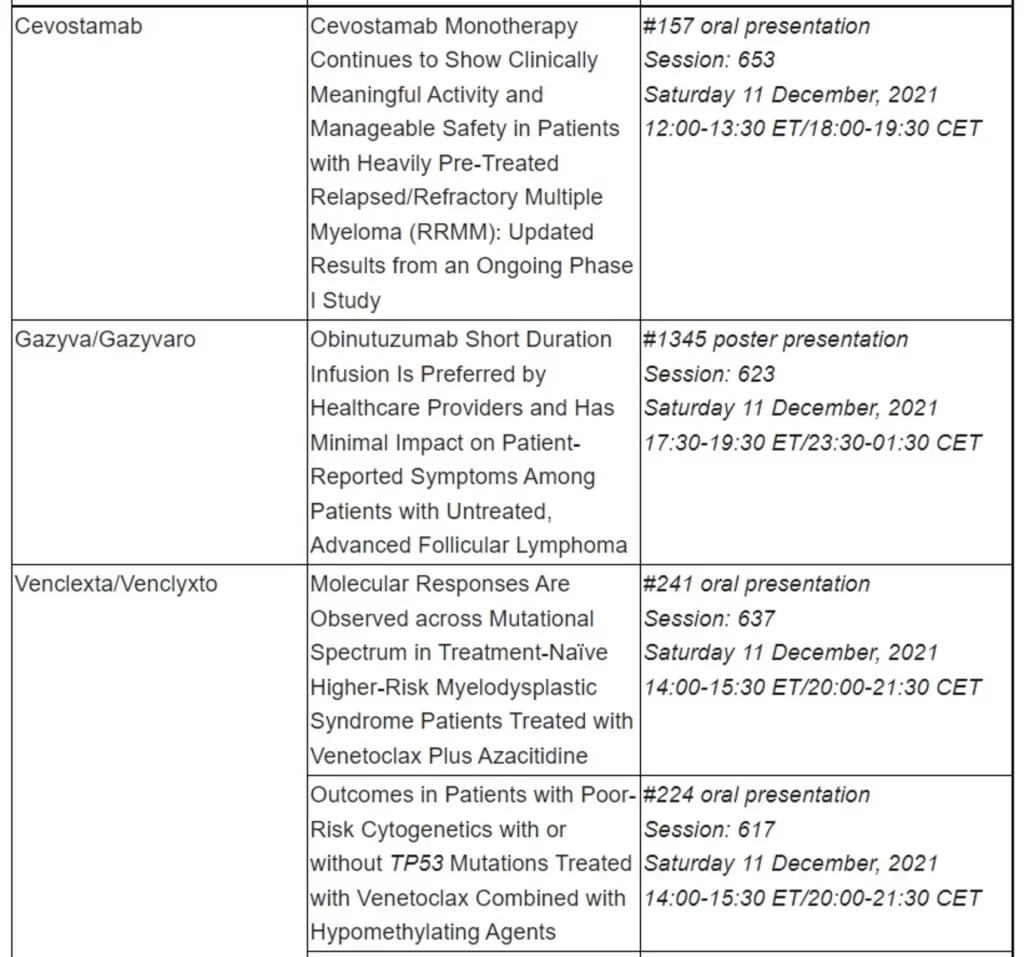
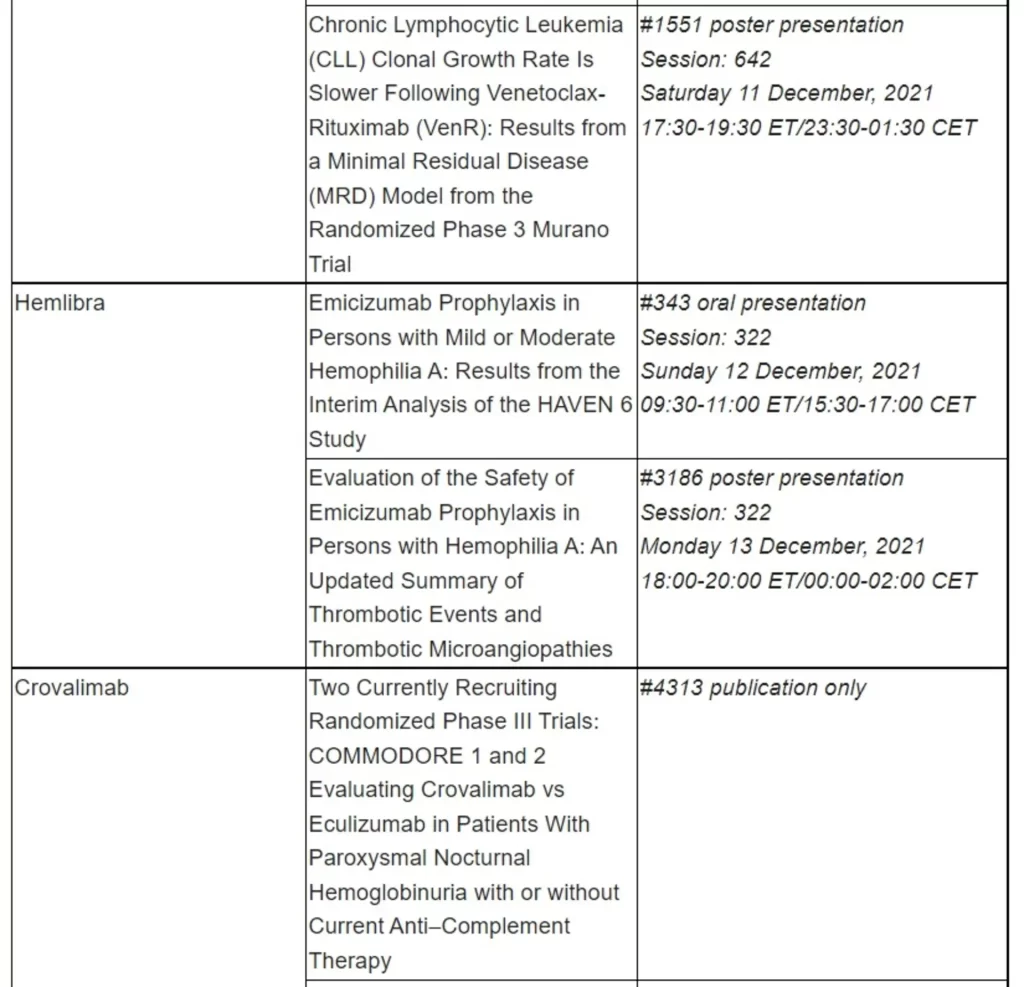

Roche has been developing medicines for people with malignant and non-malignant blood diseases for over 20 years; our experience and knowledge in this therapeutic area runs deep. Today, we are investing more than ever in our effort to bring innovative treatment options to patients across a wide range of haematologic diseases. Our approved medicines include MabThera®/Rituxan® (rituximab), Gazyva®/Gazyvaro® (obinutuzumab), Polivy® (polatuzumab vedotin), Venclexta®/Venclyxto® (venetoclax) in collaboration with AbbVie, and Hemlibra® (emicizumab). Our pipeline of investigational haematology medicines includes T-cell engaging bispecific antibodies, glofitamab and mosunetuzumab, targeting both CD20 and CD3, and cevostamab, targeting both FcRH5 and CD3; Tecentriq® (atezolizumab), a monoclonal antibody designed to bind with PD-L1; and crovalimab, an anti-C5 antibody engineered to optimise complement inhibition. Our scientific expertise, combined with the breadth of our portfolio and pipeline, also provides a unique opportunity to develop combination regimens that aim to improve the lives of patients even further.
Roche is a global pioneer in pharmaceuticals and diagnostics focused on advancing science to improve people’s lives. The combined strengths of pharmaceuticals and diagnostics under one roof have made Roche the leader in personalised healthcare – a strategy that aims to fit the right treatment to each patient in the best way possible.
Roche is the world’s largest biotech company, with truly differentiated medicines in oncology, immunology, infectious diseases, ophthalmology and diseases of the central nervous system. Roche is also the world leader in in vitro diagnostics and tissue-based cancer diagnostics, and a frontrunner in diabetes management.
Founded in 1896, Roche continues to search for better ways to prevent, diagnose and treat diseases and make a sustainable contribution to society. The company also aims to improve patient access to medical innovations by working with all relevant stakeholders. More than thirty medicines developed by Roche are included in the World Health Organization Model Lists of Essential Medicines, among them life-saving antibiotics, antimalarials and cancer medicines. Moreover, for the twelfth consecutive year, Roche has been recognised as one of the most sustainable companies in the Pharmaceuticals Industry by the Dow Jones Sustainability Indices (DJSI).
The Roche Group, headquartered in Basel, Switzerland, is active in over 100 countries and in 2020 employed more than 100,000 people worldwide. In 2020, Roche invested CHF 12.2 billion in R&D and posted sales of CHF 58.3 billion. Genentech, in the United States, is a wholly owned member of the Roche Group. Roche is the majority shareholder in Chugai Pharmaceutical, Japan
Gavreto is the first and only precision medicine approved in the EU for first-line treatment of people with RET fusion-positive advanced NSCLC
Conditional approval is based on results from the phase I/II ARROW study, in which Gavreto led to durable responses in people with RET fusion-positive advanced NSCLC
Basel, 19 November 2021 - Roche (SIX: RO, ROG; OTCQX: RHHBY) today announced that the European Commission (EC) has granted conditional marketing authorisation for Gavreto® (pralsetinib) as a monotherapy for the treatment of adults with rearranged during transfection (RET) fusion-positive advanced non-small cell lung cancer (NSCLC) not previously treated with a RET inhibitor. Gavreto is the first and only precision medicine approved in the European Union (EU) for the first-line treatment of people with RET fusion-positive advanced NSCLC.1
“Today’s approval represents an important step forward in delivering precision medicine to people with RET fusion-positive advanced non-small cell lung cancer, for whom treatment options have historically been limited," said Levi Garraway, M.D., Ph.D., Roche’s Chief Medical Officer and Head of Global Product Development. “By using cancer genomic profiling upfront, healthcare professionals may identify specific genetic alterations that predict clinical benefit of targeted treatment options like Gavreto in the first-line setting.”
The approval is based on results of the ongoing phase I/II ARROW study, in which Gavreto led to durable responses in people with advanced RET fusion-positive NSCLC.2 In 75 treatment-naïve patients, Gavreto demonstrated an overall response rate (ORR) of 72.0% (95% CI: 60.4%, 81.8%), and median duration of response (DOR) was not reached (NR) (95% CI: 9.0 months, NR).2 In 136 patients who had previously received platinum-based chemotherapy, Gavreto demonstrated an ORR of 58.8% (95% CI: 50.1%, 67.2%), and median DOR was 22.3 months (95% CI: 15.1 months, NR).2 Gavreto was also generally well-tolerated, with a low rate of treatment discontinuation; common grade 3-4 adverse reactions were neutropenia (reported in 20.1% of patients), anaemia (17.6%) and hypertension (16.1%).2
Approximately 37,500 people are diagnosed with RET fusion-positive NSCLC worldwide each year; the disease often affects people with minimal to no history of smoking, and who are typically younger than the average person diagnosed with lung cancer.3,4,5 Roche is committed to providing a tailored treatment option for every person with lung cancer, no matter how rare or difficult-to-treat their type of disease. Gavreto in RET fusion-positive advanced NSCLC, along with Alecensa® (alectinib) in ALK-positive advanced NSCLC and Rozlytrek® (entrectinib) in ROS1-positive advanced NSCLC, is part of Roche’s growing portfolio of precision medicines. Together, they offer personalised treatment options for almost one in ten people with advanced NSCLC, and biomarker testing is the most effective way to identify those people who may benefit.6
Beyond NSCLC, RET alterations are also key disease drivers in other cancer types, such as thyroid cancers. Gavreto has shown activity across multiple solid tumour types, reflecting tumour-agnostic potential.7 It is approved by the U.S. Food and Drug Administration (FDA) for the treatment of adults with metastatic RET fusion-positive NSCLC, and for the treatment of adult and paediatric patients 12 years of age and older with advanced RET-altered thyroid cancers. Gavreto is also approved in Canada, mainland China and Switzerland. In the EU, a submission for RET-altered thyroid cancers is planned. Regulatory submissions for advanced RET fusion-positive NSCLC and RET-altered thyroid cancers are also underway in multiple countries worldwide.
Blueprint Medicines and Roche are co-developing Gavreto globally, with the exception of certain territories in Asia, including China.* Blueprint Medicines and Genentech, a wholly owned member of the Roche Group, are commercialising Gavreto in the US and Roche has exclusive commercialisation rights for Gavreto outside of the US, with the exception of certain territories in Asia, including China.*
ARROW is an ongoing phase I/II, open-label, first-in-human study designed to evaluate the safety, tolerability and efficacy of Gavreto, administered orally in people with rearranged during transfection (RET) fusion-positive non-small cell lung cancer (NSCLC), RET-mutant medullary thyroid cancer, RET fusion-positive thyroid cancer and other RET-altered solid tumours. ARROW is being conducted at multiple sites across the United States, Europe and Asia.
RET gene alterations, such as fusions and mutations, are key disease drivers in many types of cancer, including non-small cell lung cancer (NSCLC) and several types of thyroid cancer. There are approximately 2.21 million cases of lung cancer diagnosed each year worldwide,3 of which approximately 1.8 million are NSCLC and RET fusions are present in approximately 1-2% of these patients,4,5 meaning RET fusion-positive NSCLC affects up to 37,500 people each year. Additionally, approximately 10-20% of people with papillary thyroid cancer (the most common type of thyroid cancer) have RET fusion-positive tumours,9 and roughly 90% of people with advanced medullary thyroid cancer (a less prevalent form of thyroid cancer) carry RET mutations.10 Oncogenic RET fusions also are observed at low frequencies in other cancers, including cholangiocarcinoma, colorectal, neuroendocrine, ovarian, pancreatic and thymus cancers.
Gavreto is a once-daily, oral precision medicine designed to selectively target rearranged during transfection (RET) alterations, including fusions and mutations, regardless of the tissue of origin. Preclinical data have shown that Gavreto inhibits primary RET fusions and mutations that cause cancer in subsets of patients, as well as secondary RET mutations predicted to drive resistance to treatment. Blueprint Medicines and Roche are co-developing Gavreto for the treatment of people with various types of RET-altered cancers.
Lung cancer is a major area of focus and investment for Roche, and we are committed to developing new approaches, medicines and tests that can help people with this deadly disease. Our goal is to provide an effective treatment option for every person diagnosed with lung cancer. We currently have six approved medicines to treat certain kinds of lung cancer, and a pipeline of investigational medicines to target the most common genetic drivers of lung cancer, or to boost the immune system to combat the disease.
Roche is a global pioneer in pharmaceuticals and diagnostics focused on advancing science to improve people’s lives. The combined strengths of pharmaceuticals and diagnostics under one roof have made Roche the leader in personalised healthcare – a strategy that aims to fit the right treatment to each patient in the best way possible.
Roche is the world’s largest biotech company, with truly differentiated medicines in oncology, immunology, infectious diseases, ophthalmology and diseases of the central nervous system. Roche is also the world leader in in vitro diagnostics and tissue-based cancer diagnostics, and a frontrunner in diabetes management.
Founded in 1896, Roche continues to search for better ways to prevent, diagnose and treat diseases and make a sustainable contribution to society. The company also aims to improve patient access to medical innovations by working with all relevant stakeholders. More than thirty medicines developed by Roche are included in the World Health Organization Model Lists of Essential Medicines, among them life-saving antibiotics, antimalarials and cancer medicines. Moreover, for the twelfth consecutive year, Roche has been recognised as one of the most sustainable companies in the Pharmaceuticals Industry by the Dow Jones Sustainability Indices (DJSI).
The Roche Group, headquartered in Basel, Switzerland, is active in over 100 countries and in 2020 employed more than 100,000 people worldwide. In 2020, Roche invested CHF 12.2 billion in R&D and posted sales of CHF 58.3 billion. Genentech, in the United States, is a wholly owned member of the Roche Group. Roche is the majority shareholder in Chugai Pharmaceutical, Japan