FIREFISH Part 2 study showed treatment with Evrysdi helped babies stay free of permanent ventilation, sit without support and improve across a range of motor milestones
Evrysdi has proven efficacy in adults, children and babies two months and older with over 4,000 patients treated to date
SMA is the leading genetic cause of death in infants
Basel, 29 July 2021 – Roche (SIX: RO, ROG; OTCQX: RHHBY) today announced that the New England Journal of Medicine (NEJM) has published data from FIREFISH Part 2, a pivotal global study evaluating the efficacy and safety of Evrysdi® (risdiplam) in babies aged 1-7 months old with symptomatic Type 1 spinal muscular atrophy (SMA). The study met its primary endpoint with 29% of infants (12/41) sitting without support for at least five seconds* by month 12, a milestone not seen in the natural course of the disease. Safety for Evrysdi in the FIREFISH Part 2 study was consistent with its known safety profile.
“Without treatment, babies with Type 1 SMA are unlikely to survive beyond two years of age,” said Professor Laurent Servais, M.D., Ph.D., FIREFISH investigator and Professor of Paediatric Neuromuscular Diseases at the MDUK Oxford Neuromuscular Centre. “Important motor milestones, such as sitting, rolling over and swallowing, are the fundamental building blocks that can help these babies achieve optimal outcomes with Evrysdi, potentially reducing the need for ventilation and increasing the rate of survival.’
At the time of the data analysis, the median duration of treatment with Evrysdi was 15.2 months and the median age was 20.7 months. At month 12, 93% (38/41) of infants were alive and 85% (35/41) were free from permanent ventilation. Without treatment, the median age of death or permanent ventilation was 13.5 months in a natural history cohort. Ninety percent (37/41) had a CHOP-INTEND** score increase of at least 4 points, with 56% (23/41) achieving a score above 40; the median increase was 20 points.
In addition, the study met one of its secondary endpoints with 78% (32/41) of infants classified as HINE-2*** responders, which evaluated motor function through head control, sitting, voluntary grasp, ability to kick, rolling, crawling, standing and walking. Infants were classified as HINE-2 responders if more motor milestones showed improvement than worsened.
“These data published in the New England Journal of Medicine validate results from Part 1 of the FIREFISH study that showed Evrysdi can help babies with SMA reach the significant milestone of sitting without support for at least five seconds,” said Levi Garraway, M.D., Ph. D., Roche’s Chief Medical Officer and Head of Global Product Development. “These results have been further confirmed in the recently presented 24 month data showing Evrysdi continued to improve motor function, doubling the number of babies able to sit without support from month 12. We will continue to work closely with governments and the SMA community to bring Evrysdi to as many people as possible.”
Safety for Evrysdi in the FIREFISH Part 2 study was consistent with its known safety profile. The most common adverse events were upper respiratory tract infection (68%), pneumonia (39%), pyrexia (39%), constipation (20%), diarrhoea (10%) and maculopapular rash (10%). The most common serious adverse events were pneumonia (32%), bronchiolitis (5%), hypotonia (5%) and respiratory failure (5%). Three infants experienced fatal complications of their disease within the first three months of treatment. None of these were attributed by the investigator as related to Evrysdi.
In February 2021, 12 month results from the dose finding Part 1 of the FIREFISH study were published in NEJM.
Roche leads the clinical development of Evrysdi as part of a collaboration with the SMA Foundation and PTC Therapeutics.
*As assessed by the Gross Motor Scale of the Bayley Scales of Infant and Toddler Development Third Edition (BSID-III)
**Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders
***Hammersmith Infant Neurological Examination 2
Evrysdi is a survival motor neuron 2 (SMN2) splicing modifier designed to treat SMA caused by mutations in chromosome 5q that lead to SMN protein deficiency. Evrysdi is administered daily at home in liquid form by mouth or by feeding tube.
Evrysdi is designed to treat SMA by increasing and sustaining the production of the survival motor neuron (SMN) protein. SMN protein is found throughout the body and is critical for maintaining healthy motor neurons and movement.
Evrysdi was granted orphan designation by the European Medicines Agency (EMA) in 2019, PRIME designation by the EMA in 2018 and Orphan Drug Designation by the U.S Food and Drug Administration in 2017. Evrysdi has been approved in 54 countries and submitted in a further 33 countries.
Evrysdi is currently being evaluated in four multicentre trials in people with SMA:
FIREFISH (NCT02913482) – an open-label, two-part pivotal clinical trial in infants with Type 1 SMA. Part 1 was a dose-escalation study in 21 infants with the primary objective of assessing the safety profile of Evrysdi in infants and determining the dose for Part 2. Part 2 is a pivotal, single-arm study of Evrysdi in 41 infants with Type 1 SMA treated for 2 years, followed by an open-label extension. Enrolment for Part 2 was completed in November 2018. The primary objective of Part 2 was to assess efficacy as measured by the proportion of infants sitting without support after 12 months of treatment, as assessed by the Gross Motor Scale of the Bayley Scales of Infant and Toddler Development – Third Edition (BSID-III) (defined as sitting without support for 5 seconds). The study met its primary endpoint.
SUNFISH (NCT02908685) – SUNFISH is a two part, double-blind, placebo controlled pivotal study in people aged 2-25 years with Types 2 or 3 SMA. Part 1 (n=51) determined the dose for the confirmatory Part 2. Part 2 (n=180) evaluated motor function using the total score of Motor Function Measure 32 (MFM-32) at 12 months. MFM-32 is a validated scale used to evaluate fine and gross motor function in people with neurological disorders, including SMA. The study met its primary endpoint.
JEWELFISH (NCT03032172) – an open-label exploratory trial designed to assess the safety, tolerability, pharmacokinetics and pharmacodynamics in people with SMA aged 6 months to 60 years (inclusion criteria) who received other investigational or approved SMA therapies for at least 90 days prior to receiving Evrysdi. The study has completed recruitment (n=174). RAINBOWFISH (NCT03779334) – an open-label, single-arm, multicentre study, investigating the efficacy, safety, pharmacokinetics and pharmacodynamics of risdiplam in babies (~n=25), from birth to six weeks of age (at first dose) with genetically diagnosed SMA who are not yet presenting with symptoms. The study is currently recruiting.
SMA is a severe, progressive neuromuscular disease that can be fatal. It affects approximately one in 10,000 babies and is the leading genetic cause of infant mortality. SMA is caused by a mutation of the survival motor neuron 1 (SMN1) gene, which leads to a deficiency of SMN protein. This protein is found throughout the body and is essential to the function of nerves that control muscles and movement. Without it, nerve cells cannot function correctly, leading to muscle weakness over time. Depending on the type of SMA, an individual’s physical strength and their ability to walk, eat or breathe can be significantly diminished or lost.
Neuroscience is a major focus of research and development at Roche. Our goal is to pursue groundbreaking science to develop new treatments that help improve the lives of people with chronic and potentially devastating diseases.
Roche is investigating more than a dozen medicines for neurological disorders, including multiple sclerosis, neuromyelitis optica spectrum disorder, Alzheimer’s disease, Huntington’s disease, Parkinson’s disease, Duchenne muscular dystrophy and autism spectrum disorder. Together with our partners, we are committed to pushing the boundaries of scientific understanding to solve some of the most difficult challenges in neuroscience today.
Roche is a global pioneer in pharmaceuticals and diagnostics focused on advancing science to improve people’s lives. The combined strengths of pharmaceuticals and diagnostics under one roof have made Roche the leader in personalised healthcare – a strategy that aims to fit the right treatment to each patient in the best way possible.
Roche is the world’s largest biotech company, with truly differentiated medicines in oncology, immunology, infectious diseases, ophthalmology and diseases of the central nervous system. Roche is also the world leader in in vitro diagnostics and tissue-based cancer diagnostics, and a frontrunner in diabetes management.
Founded in 1896, Roche continues to search for better ways to prevent, diagnose and treat diseases and make a sustainable contribution to society. The company also aims to improve patient access to medical innovations by working with all relevant stakeholders. More than thirty medicines developed by Roche are included in the World Health Organization Model Lists of Essential Medicines, among them life-saving antibiotics, antimalarials and cancer medicines. Moreover, for the twelfth consecutive year, Roche has been recognised as one of the most sustainable companies in the Pharmaceuticals Industry by the Dow Jones Sustainability Indices (DJSI).
The Roche Group, headquartered in Basel, Switzerland, is active in over 100 countries and in 2020 employed more than 100,000 people worldwide. In 2020, Roche invested CHF 12.2 billion in R&D and posted sales of CHF 58.3 billion. Genentech, in the United States, is a wholly owned member of the Roche Group. Roche is the majority shareholder in Chugai Pharmaceutical, Japan
Across four phase III studies, approximately half of patients receiving faricimab could extend treatment time to every four months – the first time this level of durability has been achieved in phase III nAMD and DME studies
If approved, faricimab would be the first and only medicine designed to target two distinct pathways that drive retinal diseases that can cause vision loss
The European Medicines Agency has also validated the faricimab Marketing Authorisation Application submission in nAMD and DME
Basel, 29 July 2021 - Roche (SIX: RO, ROG; OTCQX: RHHBY) today announced that the U.S. Food and Drug Administration (FDA) has accepted the company’s Biologics License Application (BLA), under Priority Review, for faricimab for the treatment of neovascular or “wet” age-related macular degeneration (nAMD) and diabetic macular edema (DME). The FDA has also accepted the company’s submission for diabetic retinopathy.
Faricimab will be the first and only bispecific antibody designed for the eye, if approved. It targets two distinct pathways – via angiopoietin-2 (Ang-2) and vascular endothelial growth factor-A (VEGF-A) – that drive a number of retinal conditions that can cause vision loss.1
“If approved, faricimab would be the first in a new class of eye medicines targeting two key pathways that drive retinal disorders, with the potential to offer durable vision outcomes with fewer eye injections than the current standard of care,” said Levi Garraway, M.D., Ph.D., Roche’s Chief Medical Officer and Head of Global Product Development. “Therefore, we hope faricimab will become a new treatment option for millions of people living with nAMD and DME.”
Neovascular AMD and DME are two leading causes of vision loss among adults worldwide.2 The BLA submission is based on positive results across four phase III studies in nAMD and DME. The studies consistently showed that faricimab, given at intervals of up to four months, offered non-inferior vision gains compared with aflibercept, given every two months. Approximately half of people eligible for extended dosing with faricimab were able to be treated every four months in the first year in the TENAYA and LUCERNE studies in nAMD and the YOSEMITE and RHINE studies in DME. Faricimab is the first injectable eye medicine to achieve this length of time between treatments in phase III studies for nAMD and DME. Furthermore, approximately three-quarters of people eligible for extended dosing with faricimab were able to be treated every three months or longer in the first year. Faricimab was generally well-tolerated in all four studies, with no new or unexpected safety signals identified.3,4
Roche also has long-term extension studies underway for faricimab. These include AVONELLE X, an extension study of TENAYA and LUCERNE evaluating the long-term safety and efficacy of faricimab in nAMD, and RHONE X, an extension study of YOSEMITE and RHINE evaluating the long-term safety and efficacy of faricimab in DME.5,6 Additionally, the COMINO and BALATON trials are also underway, evaluating the efficacy and safety of faricimab in people with macular edema secondary to two types of retinal vein occlusion (RVO): central RVO and branch RVO.7,8
The European Medicines Agency has also validated the faricimab Marketing Authorisation Application for the treatment of nAMD and DME.
TENAYA (NCT03823287) and LUCERNE (NCT03823300) are two identical, randomised, multicentre, double-masked, global phase III studies evaluating the efficacy and safety of faricimab compared to aflibercept in 1,329 people living with neovascular age-related macular degeneration (671 in TENAYA and 658 in LUCERNE). The studies each have two treatment arms: faricimab 6.0 mg administered at fixed intervals of every two, three, or four months, selected based on objective assessment of disease activity at weeks 20 and 24; and aflibercept 2.0 mg administered at fixed two-month intervals. In both arms, sham injections were administered at study visits when treatment injections were not scheduled, to maintain the masking of investigators and participants.
The primary endpoint of the studies is the average change in best-corrected visual acuity (BCVA) score (the best distance vision a person can achieve – including with correction such as glasses – when reading letters on an eye chart) from baseline through week 48. Secondary endpoints include: safety; the percentage of participants in the faricimab arm receiving treatment every two, three and four months; the percentage of participants achieving a gain, and the percentage avoiding a loss, of 15 letters or more in BCVA from baseline over time; and change in central subfield thickness (CST) from baseline over time.
Both studies met their primary endpoint, with faricimab consistently shown to offer non-inferior visual acuity gains to aflibercept. In TENAYA and LUCERNE, the average vision gains from baseline in the faricimab arms were +5.8 and +6.6 letters, respectively, compared to +5.1 and +6.6 letters in the aflibercept arms.
The studies also measured the proportion of people in the faricimab arm that were treated on dosing schedules of every three or four months during the first year. Importantly, 46% (n=144/315) of patients in TENAYA and 45% (n=142/316) in LUCERNE were able to be treated every four months in the first year. An additional 34% (n=107/315) of patients in TENAYA and 33% (n=104/316) in LUCERNE were able to be treated every three months. Combined, nearly 80% of faricimab-treated patients were able to go three months or longer between treatments during the first year. In both studies, faricimab given at intervals of up to four months offered reductions in CST comparable to aflibercept given every two months. Faricimab was generally well-tolerated in both studies, with no new or unexpected safety signals identified.
YOSEMITE (NCT03622580) and RHINE (NCT03622593) are two identical, randomised, multicentre, double-masked, global phase III studies evaluating the efficacy and safety of faricimab compared to aflibercept in 1,891 people with diabetic macular edema (940 in YOSEMITE and 951 in RHINE). The studies each have three treatment arms: faricimab 6.0 mg administered at personalised treatment intervals (PTI) of up to four months; faricimab 6.0 mg administered at fixed two-month intervals; and aflibercept 2.0 mg administered at fixed two-month intervals. In all three arms, sham injections were administered at study visits when treatment injections were not scheduled, to maintain the masking of investigators and participants.
The primary endpoint of the studies is the average change in best-corrected visual acuity (BCVA) score from baseline at one year. Secondary endpoints include: safety; the percentage of participants in the personalised dosing arm receiving treatment every one, two, three and four months, at week 52; the percentage of participants achieving a two-step or greater improvement from baseline in diabetic retinopathy severity at week 52; the percentage of participants achieving a gain, and the percentage avoiding a loss of 15 letters or more in BCVA from baseline over time and change in central subfield thickness (CST) from baseline over time.
Both studies met their primary endpoint with faricimab consistently shown to offer non-inferior visual acuity gains to aflibercept. In YOSEMITE, the average vision gains from baseline were +11.6 and +10.7 eye chart letters in the faricimab PTI and two-month arms, respectively, and +10.9 letters in the aflibercept arm. In RHINE, the average vision gains from baseline were +10.8 and +11.8 letters in the faricimab PTI and two-month arms, respectively, and +10.3 letters in the aflibercept arm.
A secondary endpoint in both studies measured the proportion of people in the faricimab PTI arm that achieved dosing schedules of every three or four months at the end of the first year. Importantly, 53% (n=151/286) of faricimab PTI patients in YOSEMITE and 51% (n=157/308) in RHINE achieved four-month dosing at one year. An additional 21% (n=60/286) of faricimab PTI patients in YOSEMITE and 20% (n=62/308) in RHINE achieved three-month dosing. Combined, more than 70% of faricimab PTI patients were able to go three months or longer between treatments at the end of the first year. In both studies, faricimab given at intervals of up to four months demonstrated greater reductions in CST compared to aflibercept given every two months. Faricimab was generally well-tolerated in both studies, with no new or unexpected safety signals identified.
Age-related macular degeneration (AMD) is a condition that affects the part of the eye that provides sharp, central vision needed for activities like reading.9 Neovascular or “wet” AMD (nAMD) is an advanced form of the disease that can cause rapid and severe vision loss.10 It develops when new and abnormal blood vessels grow uncontrolled under the macula, causing swelling, bleeding and/or fibrosis.11 Worldwide, around 20 million people are living with nAMD – the leading cause of vision loss in people over the age of 60 – and the condition will affect even more people around the world as the global population ages.
Affecting around 21 million people globally, diabetic macular edema (DME) is a vision-threatening complication of diabetic retinopathy (DR).14 DR occurs when damage to blood vessels and the formation of new blood vessels cause blood and/or fluid to leak into the retina – a part of the eye that sends information to the brain, enabling sight.15 This leads to swelling, as well as blockage of blood supply to some areas of the retina.16 DME occurs when the damaged blood vessels leak into and cause swelling in the macula – the central area of the retina responsible for the sharp vision needed for reading and driving.15,17 The number of people with DME is expected to grow as the prevalence of diabetes increases.18 The condition is associated with blindness when left untreated and decreased quality of life.19 There remains a significant unmet need for more effective, longer-lasting therapies for people with DME.
Faricimab is the first investigational bispecific antibody designed for the eye. It targets two distinct pathways – via angiopoietin-2 (Ang-2) and vascular endothelial growth factor-A (VEGF-A) – that drive a number of retinal conditions.1 Ang-2 and VEGF-A contribute to vision loss by destabilising blood vessels, causing new leaky blood vessels to form and increasing inflammation.2 By simultaneously blocking both pathways involving Ang-2 and VEGF-A, faricimab is designed to stabilise blood vessels, potentially improving vision outcomes for longer for people living with retinal conditions.
Roche is focused on saving people’s eyesight from the leading causes of vision loss through pioneering therapies. Through our innovation in the scientific discovery of new potential drug targets, personalised healthcare, molecular engineering, biomarkers and continuous drug delivery, we strive to design the right therapies for the right patients.
We have the broadest retina pipeline in Ophthalmology, covering early and late stage products, which is led by science and informed by insights from people with eye diseases. Our late stage pipeline includes two potential first-of-a-kind treatments, Port Delivery System with ranibizumab (PDS) and faricimab, which are being evaluated in a number of retinal conditions including neovascular age-related macular degeneration, diabetic macular edema and diabetic retinopathy. PDS is an investigational, permanent refillable eye implant that continuously delivers a customised formulation of ranibizumab over a period of months, potentially reducing the treatment burden associated with frequent eye injections.20 Faricimab is the first investigational bispecific antibody designed for the eye.1 It targets two distinct pathways – via angiopoietin-2 (Ang-2) and vascular endothelial growth factor-A (VEGF-A) – that drive a number of retinal conditions, to stabilise blood vessels, potentially improving vision outcomes for longer.1,2 Our early stage pipeline includes gene therapies and treatments for geographic atrophy and other vision-threatening diseases, including rare and inherited conditions.
Applying our extensive experience, we have already brought breakthrough ophthalmic treatments to people living with vision loss through Lucentis®️* (ranibizumab injection), the first treatment approved to improve vision in people with certain retinal conditions.
Roche is a global pioneer in pharmaceuticals and diagnostics focused on advancing science to improve people’s lives. The combined strengths of pharmaceuticals and diagnostics under one roof have made Roche the leader in personalised healthcare – a strategy that aims to fit the right treatment to each patient in the best way possible.
Roche is the world’s largest biotech company, with truly differentiated medicines in oncology, immunology, infectious diseases, ophthalmology and diseases of the central nervous system. Roche is also the world leader in in vitro diagnostics and tissue-based cancer diagnostics, and a frontrunner in diabetes management.
Founded in 1896, Roche continues to search for better ways to prevent, diagnose and treat diseases and make a sustainable contribution to society. The company also aims to improve patient access to medical innovations by working with all relevant stakeholders. More than thirty medicines developed by Roche are included in the World Health Organization Model Lists of Essential Medicines, among them life-saving antibiotics, antimalarials and cancer medicines. Moreover, for the twelfth consecutive year, Roche has been recognised as one of the most sustainable companies in the Pharmaceuticals Industry by the Dow Jones Sustainability Indices (DJSI).
The Roche Group, headquartered in Basel, Switzerland, is active in over 100 countries and in 2020 employed more than 100,000 people worldwide. In 2020, Roche invested CHF 12.2 billion in R&D and posted sales of CHF 58.3 billion. Genentech, in the United States, is a wholly owned member of the Roche Group. Roche is the majority shareholder in Chugai Pharmaceutical, Japan
ENSPRYNG is the first and only treatment approved for both adults and adolescents in the EU with AQP4-IgG seropositive NMOSD
ENSPRYNG can be used as a monotherapy or in combination with immunosuppressive therapy to reduce relapses and prevent permanent disability
In Phase III studies, ENSPRYNG significantly reduced the number and severity of relapses in people with AQP4-IgG seropositive NMOSD
Basel, 28 June 2021 - Roche (SIX: RO, ROG; OTCQX: RHHBY) announced today that the European Commission (EC) has approved ENSPRYNG® (satralizumab) for the treatment of adults and adolescents from 12 years of age living with anti-aquaporin-4 antibody (AQP4-IgG) seropositive neuromyelitis optica spectrum disorder (NMOSD), as a monotherapy or in combination with immunosuppressive therapy (IST). ENSPRYNG is the first and only NMOSD treatment that is administered subcutaneously every four weeks, allowing home-dosing after appropriate training.
“An NMOSD relapse can be devastating, causing permanent neurological damage and disability that accumulates with subsequent relapses, which is why our goal is to prevent them,” said Prof. Dr. Friedemann Paul, Professor of Clinical Neuroimmunology, Charité Universitätsmedizin Berlin. “With the approval of ENSPRYNG, we now have a treatment option with a favourable safety profile that significantly reduces relapses in AQP4-IgG seropositive adults and adolescents after their first NMOSD attack or in more advanced disease, either as a monotherapy or in combination with IST. Importantly, people with NMOSD now have the flexibility to administer treatment at home, which may alleviate the need to travel for hospital appointments.”
The EC approval is supported by results from two Phase III studies, in which ENSPRYNG showed robust and sustained efficacy in reducing the risk of relapse in people with AQP4-IgG seropositive NMOSD. AQP4-IgG are present in around 70-80% of people with NMOSD, who tend to experience a more severe disease course compared to those not expressing AQP4-IgG antibodies.
“We thank the NMOSD community for their partnership and are delighted that ENSPRYNG will be available to people in the EU who until now had limited, accessible treatment options,” said Levi Garraway, M.D., Ph.D., Chief Medical Officer and Head of Global Product Development. “Building on our growing scientific understanding of neuroimmunological conditions, we are confident ENSPRYNG can transform how people with NMOSD are treated by fitting into their day-to-day lives.”
ENSPRYNG is the first and only approved medicine for NMOSD in the EU designed to bind to and block the interleukin-6 (IL-6) receptor, a central driver of the inflammation associated with NMOSD. The treatment was designed by Chugai, a member of the Roche Group, using novel recycling antibody technology. When compared to conventional antibodies, ENSPRYNG’s recycling antibody technology enables the medicine to remain in the bloodstream for a longer period of time and bind repeatedly to its target (the IL-6 receptor) – maximally sustaining IL-6 suppression in a chronic disease like NMOSD and enabling subcutaneous dosing every four weeks.
Roche is working closely with reimbursement and health technology assessment bodies in EU member states to provide access to ENSPRYNG for people who may benefit from this treatment option as soon as possible.
ENSPRYNG has been investigated in two pivotal Phase III studies in neuromyelitis optica spectrum disorder (NMOSD), with the primary endpoint of both studies being time to first protocol-defined relapse (PDR) adjudicated by an independent review committee in the double-blind period.
The Phase III SAkuraStar study evaluated the efficacy and safety of ENSPRYNG monotherapy administered to adults with NMOSD. In the anti-aquaporin-4 antibody (AQP4-IgG) seropositive subgroup, 83% treated with ENSPRYNG remained relapse free at 48 weeks, compared with 55% of those treated with placebo. At 96 weeks, 77% of those treated with ENSPRYNG remained relapse free, compared with 41% with placebo.
The Phase III SAkuraSky study evaluated the efficacy and safety of ENSPRYNG in combination with baseline immunosuppressive therapy in adults and adolescents with NMOSD. Overall, 92% of AQP4-IgG seropositive participants receiving ENSPRYNG in combination with IST remained relapse free at 48 and 96 weeks, compared with 60% and 53% with placebo, respectively.
ENSPRYNG showed a favourable safety and tolerability profile in the Phase III studies. The most common adverse reactions observed in the safety population were: headache, arthralgia, white blood cell count decrease, hyperlipidaemia and injection-related reactions.
NMOSD is a rare, lifelong and debilitating autoimmune condition of the central nervous system that primarily damages the optic nerve(s) and spinal cord, causing permanent blindness, muscle weakness and paralysis. People with NMOSD experience unpredictable, severe relapses directly causing cumulative, permanent, neurological damage and disability. In some cases, relapse can result in death. NMOSD affects over 10,000 people in Europe, up to 15,000 people in the US and approximately 200,000 people worldwide. NMOSD can affect individuals of any age, race and gender, but is most common among women in their 30s and 40s, and appears to occur at higher rates in people of African or Asian background.
NMOSD is commonly associated with pathogenic antibodies (AQP4-IgG) that target and damage a specific cell type, called astrocytes, resulting in inflammatory lesions of the optic nerve(s), spinal cord and brain. AQP4-IgG antibodies are detectable in the blood serum of around 70-80% of people with NMOSD.
Although most cases of NMOSD can be confirmed through diagnostic tests, people living with the condition are still frequently misdiagnosed with multiple sclerosis. This is due to overlapping characteristics of the two disorders, including a higher prevalence in women, similar symptoms and the fact that people can experience relapses in both conditions.
ENSPRYNG, which was designed by Chugai, a member of the Roche Group, is a humanised monoclonal antibody that targets interleukin-6 (IL-6) receptor activity. The cytokine IL-6 is believed to be a key driver in NMOSD disease processes, triggering the inflammation cascade and leading to damage and disability. ENSPRYNG was designed using novel recycling antibody technology. When compared to conventional antibodies, ENSPRYNG’s recycling antibody technology enables the medicine to remain in the bloodstream for a longer period of time and bind repeatedly to its target (the IL-6 receptor) - maximally sustaining IL-6 suppression in a chronic disease like NMOSD and enabling subcutaneous dosing every four weeks.
Positive Phase III results for ENSPRYNG, as both monotherapy and in combination with baseline immunosuppressive therapy, suggest that IL-6 inhibition is an effective therapeutic approach for NMOSD. The Phase III clinical development programme for ENSPRYNG included two studies: SAkuraStar and SAkuraSky.
ENSPRYNG is currently approved in 54 countries, including the United States, Canada, Japan, China and EMA territory countries.
ENSPRYNG has been designated as an orphan drug in the U.S., Europe and Japan. In addition, it was granted Breakthrough Therapy Designation for the treatment of NMOSD by the FDA in December 2018, which is given to treatments that may demonstrate substantial improvement over other available options.
Neuroscience is a major focus of research and development at Roche. Our goal is to pursue groundbreaking science to develop new treatments that help improve the lives of people with chronic and potentially devastating diseases.
Roche is investigating more than a dozen medicines for neurological disorders, including multiple sclerosis, Alzheimer’s disease, Huntington’s disease, Parkinson’s disease, Duchenne muscular dystrophy and autism spectrum disorder. Together with our partners, we are committed to pushing the boundaries of scientific understanding to solve some of the most difficult challenges in neuroscience today.
Roche is a global pioneer in pharmaceuticals and diagnostics focused on advancing science to improve people’s lives. The combined strengths of pharmaceuticals and diagnostics under one roof have made Roche the leader in personalised healthcare – a strategy that aims to fit the right treatment to each patient in the best way possible.
Roche is the world’s largest biotech company, with truly differentiated medicines in oncology, immunology, infectious diseases, ophthalmology and diseases of the central nervous system. Roche is also the world leader in in vitro diagnostics and tissue-based cancer diagnostics, and a frontrunner in diabetes management.
Founded in 1896, Roche continues to search for better ways to prevent, diagnose and treat diseases and make a sustainable contribution to society. The company also aims to improve patient access to medical innovations by working with all relevant stakeholders. More than thirty medicines developed by Roche are included in the World Health Organization Model Lists of Essential Medicines, among them life-saving antibiotics, antimalarials and cancer medicines. Moreover, for the twelfth consecutive year, Roche has been recognised as one of the most sustainable companies in the Pharmaceuticals Industry by the Dow Jones Sustainability Indices (DJSI).
The Roche Group, headquartered in Basel, Switzerland, is active in over 100 countries and in 2020 employed more than 100,000 people worldwide. In 2020, Roche invested CHF 12.2 billion in R&D and posted sales of CHF 58.3 billion. Genentech, in the United States, is a wholly owned member of the Roche Group. Roche is the majority shareholder in Chugai Pharmaceutical, Japan
Every year in the US, approximately 10,000 people are diagnosed with myelodysplastic syndromes (MDS), and there remains a high unmet need for new treatment options
The designation is based on interim results from the phase Ib M15-531 study investigating Venclexta/Venclyxto plus azacitidine in people with previously untreated higher-risk MDS
This is the 11th Breakthrough Therapy Designation for Roche’s haematology medicines and the sixth for Venclexta demonstrating its potential across multiple blood cancers
Basel, 21 July 2021 - Roche (SIX: RO, ROG; OTCQX: RHHBY) today announced that Venclexta® (venetoclax) in combination with azacitidine has been granted Breakthrough Therapy Designation (BTD) by the U.S. Food and Drug Administration (FDA) for the treatment of adult patients with previously untreated intermediate, high- and very high-risk myelodysplastic syndromes (MDS) based on the revised International Prognostic Scoring System (IPSS-R). MDS are a rare group of blood cancers that gradually affect the ability of the bone marrow to produce normal blood cells.2 This can lead to weakness, frequent infections, anaemia and debilitating fatigue.3 In some cases, MDS can also progress into acute myeloid leukaemia (AML).4,5 Every year in the US, approximately 10,000 people are diagnosed with MDS, and the median survival for those with higher-risk MDS is approximately 18 months.1,3
“Higher-risk MDS is associated with poor prognosis, reduced quality of life, and limited treatment options,” said Levi Garraway, M.D., Ph.D., Roche’s Chief Medical Officer and Head of Global Product Development. “We are pleased that the FDA has granted Venclexta its sixth Breakthrough Therapy Designation in recognition of its potential to improve outcomes for people with MDS in combination with azacitidine.”
This designation was granted based on interim results from the phase Ib M15-531 study investigating Venclexta/Venclyxto plus azacitidine in people with previously untreated, higher-risk MDS. BTD is designed to accelerate the development and review of medicines intended to treat serious or life-threatening conditions with preliminary evidence that indicates they may demonstrate a substantial improvement over existing therapies. This is the 38th BTD for Roche’s portfolio of medicines, and the 11th designation for its haematology portfolio.
This most recent designation reinforces the potential of Venclexta/Venclyxto-based combinations across several blood cancers, including MDS. In the US, Venclexta has been granted six BTDs by the FDA: one for previously untreated chronic lymphocytic leukaemia (CLL), two for relapsed or refractory CLL, two for previously untreated AML, and one for MDS. Venclexta/Venclyxto is already approved in the US (as Venclexta) in combination with azacitidine, decitabine or low-dose cytarabine for the treatment of newly diagnosed AML in adults 75 years or older, or who have comorbidities that preclude use of intensive induction chemotherapy, and in the EU (as Venclyxto) in combination with hypomethylating agents, azacitidine and decitabine, for the treatment of adult patients with newly diagnosed AML who are ineligible for intensive chemotherapy. Venclexta/Venclyxto is also approved in the US and EU in combination with MabThera®/Rituxan® (rituximab) for the treatment of adult patients with CLL who have received at least one prior therapy; in combination with Gazyva®/Gazyvaro® (obinutuzumab) for the treatment of adult patients with previously untreated CLL; and as a monotherapy for the treatment of CLL in the presence of 17p deletion or TP53 mutation in people who are unsuitable for or have failed a B-cell receptor pathway inhibitor.
Venclexta/Venclyxto is being developed by AbbVie and Roche. It is jointly commercialised by AbbVie and Genentech, a member of the Roche Group, in the US, under the brand name Venclexta, and commercialised by AbbVie outside of the US.
MDS are a rare group of blood cancers that gradually affect the ability of the bone marrow to produce normal blood cells.2 This can lead to weakness, frequent infections, anaemia and debilitating fatigue.3 In some cases, MDS can also progress into acute myeloid leukaemia (AML).4,5 Every year in the US, approximately 10,000 people are diagnosed with MDS and the median survival for those with higher-risk MDS is approximately 18 months.1,3
There are several classifications of MDS – very low-risk to very high-risk – determined by the composition of the bone marrow, blood cell counts, and chromosomal alterations. Higher-risk disease is defined as intermediate, high- or very high-risk based on the revised International Prognostic Scoring System (IPSS-R), which is a risk assessment scale that uses five prognostic indicators to predict the course of a patient’s disease.7 Approximately half (45%) of patients present with higher-risk MDS, which is associated with a poorer prognosis and short life expectancy.
The M15-531 [NCT02942290] study is a phase Ib, open-label, non-randomised, multicentre, dose-finding study evaluating Venclexta®/Venclyxto® (venetoclax) in combination with azacitidine in treatment-naïve patients with higher-risk myelodysplastic syndromes (MDS) comprising a dose-escalation portion and a safety expansion portion. The primary objectives of the study are to assess the safety profile and pharmacokinetics and determine the recommended phase II dose and dosing schedule of Venclexta/Venclyxto in combination with azacitidine. The response criteria specified in the M15-531 study are based on the modified International Working Group 2006 response criteria for MDS.
Venclexta®/Venclyxto® (venetoclax) is a first-in-class targeted medicine designed to selectively bind and inhibit the B-cell lymphoma-2 (BCL-2) protein. In some blood cancers and other tumours, BCL-2 builds up and prevents cancer cells from dying or self-destructing, a process called apoptosis. Venclexta/Venclyxto blocks the BCL-2 protein and works to restore the process of apoptosis.
Venclexta/Venclyxto is being developed by AbbVie and Roche. It is jointly commercialised by AbbVie and Genentech, a member of the Roche group, in the US and commercialised by AbbVie outside of the US. Together, the companies are committed to research with Venclexta/Venclyxto, which is currently being studied in clinical trials across several types of blood cancers.
In the US, Venclexta has been granted six Breakthrough Therapy Designations by the U.S. Food and Drug Administration: one for previously untreated chronic lymphocytic leukaemia (CLL), two for relapsed or refractory CLL, two for previously untreated acute myeloid leukaemia, and one for myelodysplastic syndromes.
Roche has been developing medicines for people with malignant and non-malignant blood diseases for over 20 years; our experience and knowledge in this therapeutic area runs deep. Today, we are investing more than ever in our effort to bring innovative treatment options to patients across a wide range of haematologic diseases. Our approved medicines include MabThera®/Rituxan® (rituximab), Gazyva®/Gazyvaro® (obinutuzumab), Polivy® (polatuzumab vedotin), Venclexta®/Venclyxto® (venetoclax) in collaboration with AbbVie, and Hemlibra® (emicizumab). Our pipeline of investigational haematology medicines includes T-cell engaging bispecific antibodies, glofitamab and mosunetuzumab, targeting both CD20 and CD3, and cevostamab, targeting FcRH5 and CD3; Tecentriq® (atezolizumab), a monoclonal antibody designed to bind with PD-L1; and crovalimab, an anti-C5 antibody engineered to optimise complement inhibition. Our scientific expertise, combined with the breadth of our portfolio and pipeline, also provides a unique opportunity to develop combination regimens that aim to improve the lives of patients even further.
Roche is a global pioneer in pharmaceuticals and diagnostics focused on advancing science to improve people’s lives. The combined strengths of pharmaceuticals and diagnostics under one roof have made Roche the leader in personalised healthcare – a strategy that aims to fit the right treatment to each patient in the best way possible.
Roche is the world’s largest biotech company, with truly differentiated medicines in oncology, immunology, infectious diseases, ophthalmology and diseases of the central nervous system. Roche is also the world leader in in vitro diagnostics and tissue-based cancer diagnostics, and a frontrunner in diabetes management.
Founded in 1896, Roche continues to search for better ways to prevent, diagnose and treat diseases and make a sustainable contribution to society. The company also aims to improve patient access to medical innovations by working with all relevant stakeholders. More than thirty medicines developed by Roche are included in the World Health Organization Model Lists of Essential Medicines, among them life-saving antibiotics, antimalarials and cancer medicines. Moreover, for the twelfth consecutive year, Roche has been recognised as one of the most sustainable companies in the Pharmaceuticals Industry by the Dow Jones Sustainability Indices (DJSI).
The Roche Group, headquartered in Basel, Switzerland, is active in over 100 countries and in 2020 employed more than 100,000 people worldwide. In 2020, Roche invested CHF 12.2 billion in R&D and posted sales of CHF 58.3 billion. Genentech, in the United States, is a wholly owned member of the Roche Group. Roche is the majority shareholder in Chugai Pharmaceutical, Japan
Every year in the US, approximately 10,000 people are diagnosed with myelodysplastic syndromes (MDS), and there remains a high unmet need for new treatment options
The designation is based on interim results from the phase Ib M15-531 study investigating Venclexta/Venclyxto plus azacitidine in people with previously untreated higher-risk MDS
This is the 11th Breakthrough Therapy Designation for Roche’s haematology medicines and the sixth for Venclexta demonstrating its potential across multiple blood cancers
Basel, 21 July 2021 - Roche (SIX: RO, ROG; OTCQX: RHHBY) today announced that Venclexta® (venetoclax) in combination with azacitidine has been granted Breakthrough Therapy Designation (BTD) by the U.S. Food and Drug Administration (FDA) for the treatment of adult patients with previously untreated intermediate, high- and very high-risk myelodysplastic syndromes (MDS) based on the revised International Prognostic Scoring System (IPSS-R). MDS are a rare group of blood cancers that gradually affect the ability of the bone marrow to produce normal blood cells.2 This can lead to weakness, frequent infections, anaemia and debilitating fatigue.3 In some cases, MDS can also progress into acute myeloid leukaemia (AML).4,5 Every year in the US, approximately 10,000 people are diagnosed with MDS, and the median survival for those with higher-risk MDS is approximately 18 months.1,3
“Higher-risk MDS is associated with poor prognosis, reduced quality of life, and limited treatment options,” said Levi Garraway, M.D., Ph.D., Roche’s Chief Medical Officer and Head of Global Product Development. “We are pleased that the FDA has granted Venclexta its sixth Breakthrough Therapy Designation in recognition of its potential to improve outcomes for people with MDS in combination with azacitidine.”
This designation was granted based on interim results from the phase Ib M15-531 study investigating Venclexta/Venclyxto plus azacitidine in people with previously untreated, higher-risk MDS. BTD is designed to accelerate the development and review of medicines intended to treat serious or life-threatening conditions with preliminary evidence that indicates they may demonstrate a substantial improvement over existing therapies. This is the 38th BTD for Roche’s portfolio of medicines, and the 11th designation for its haematology portfolio.
This most recent designation reinforces the potential of Venclexta/Venclyxto-based combinations across several blood cancers, including MDS. In the US, Venclexta has been granted six BTDs by the FDA: one for previously untreated chronic lymphocytic leukaemia (CLL), two for relapsed or refractory CLL, two for previously untreated AML, and one for MDS. Venclexta/Venclyxto is already approved in the US (as Venclexta) in combination with azacitidine, decitabine or low-dose cytarabine for the treatment of newly diagnosed AML in adults 75 years or older, or who have comorbidities that preclude use of intensive induction chemotherapy, and in the EU (as Venclyxto) in combination with hypomethylating agents, azacitidine and decitabine, for the treatment of adult patients with newly diagnosed AML who are ineligible for intensive chemotherapy. Venclexta/Venclyxto is also approved in the US and EU in combination with MabThera®/Rituxan® (rituximab) for the treatment of adult patients with CLL who have received at least one prior therapy; in combination with Gazyva®/Gazyvaro® (obinutuzumab) for the treatment of adult patients with previously untreated CLL; and as a monotherapy for the treatment of CLL in the presence of 17p deletion or TP53 mutation in people who are unsuitable for or have failed a B-cell receptor pathway inhibitor.
Venclexta/Venclyxto is being developed by AbbVie and Roche. It is jointly commercialised by AbbVie and Genentech, a member of the Roche Group, in the US, under the brand name Venclexta, and commercialised by AbbVie outside of the US.
MDS are a rare group of blood cancers that gradually affect the ability of the bone marrow to produce normal blood cells.2 This can lead to weakness, frequent infections, anaemia and debilitating fatigue.3 In some cases, MDS can also progress into acute myeloid leukaemia (AML).4,5 Every year in the US, approximately 10,000 people are diagnosed with MDS and the median survival for those with higher-risk MDS is approximately 18 months.1,3
There are several classifications of MDS – very low-risk to very high-risk – determined by the composition of the bone marrow, blood cell counts, and chromosomal alterations. Higher-risk disease is defined as intermediate, high- or very high-risk based on the revised International Prognostic Scoring System (IPSS-R), which is a risk assessment scale that uses five prognostic indicators to predict the course of a patient’s disease.7 Approximately half (45%) of patients present with higher-risk MDS, which is associated with a poorer prognosis and short life expectancy.
The M15-531 [NCT02942290] study is a phase Ib, open-label, non-randomised, multicentre, dose-finding study evaluating Venclexta®/Venclyxto® (venetoclax) in combination with azacitidine in treatment-naïve patients with higher-risk myelodysplastic syndromes (MDS) comprising a dose-escalation portion and a safety expansion portion. The primary objectives of the study are to assess the safety profile and pharmacokinetics and determine the recommended phase II dose and dosing schedule of Venclexta/Venclyxto in combination with azacitidine. The response criteria specified in the M15-531 study are based on the modified International Working Group 2006 response criteria for MDS.
Venclexta®/Venclyxto® (venetoclax) is a first-in-class targeted medicine designed to selectively bind and inhibit the B-cell lymphoma-2 (BCL-2) protein. In some blood cancers and other tumours, BCL-2 builds up and prevents cancer cells from dying or self-destructing, a process called apoptosis. Venclexta/Venclyxto blocks the BCL-2 protein and works to restore the process of apoptosis.
Venclexta/Venclyxto is being developed by AbbVie and Roche. It is jointly commercialised by AbbVie and Genentech, a member of the Roche group, in the US and commercialised by AbbVie outside of the US. Together, the companies are committed to research with Venclexta/Venclyxto, which is currently being studied in clinical trials across several types of blood cancers.
In the US, Venclexta has been granted six Breakthrough Therapy Designations by the U.S. Food and Drug Administration: one for previously untreated chronic lymphocytic leukaemia (CLL), two for relapsed or refractory CLL, two for previously untreated acute myeloid leukaemia, and one for myelodysplastic syndromes.
Roche has been developing medicines for people with malignant and non-malignant blood diseases for over 20 years; our experience and knowledge in this therapeutic area runs deep. Today, we are investing more than ever in our effort to bring innovative treatment options to patients across a wide range of haematologic diseases. Our approved medicines include MabThera®/Rituxan® (rituximab), Gazyva®/Gazyvaro® (obinutuzumab), Polivy® (polatuzumab vedotin), Venclexta®/Venclyxto® (venetoclax) in collaboration with AbbVie, and Hemlibra® (emicizumab). Our pipeline of investigational haematology medicines includes T-cell engaging bispecific antibodies, glofitamab and mosunetuzumab, targeting both CD20 and CD3, and cevostamab, targeting FcRH5 and CD3; Tecentriq® (atezolizumab), a monoclonal antibody designed to bind with PD-L1; and crovalimab, an anti-C5 antibody engineered to optimise complement inhibition. Our scientific expertise, combined with the breadth of our portfolio and pipeline, also provides a unique opportunity to develop combination regimens that aim to improve the lives of patients even further.
Roche is a global pioneer in pharmaceuticals and diagnostics focused on advancing science to improve people’s lives. The combined strengths of pharmaceuticals and diagnostics under one roof have made Roche the leader in personalised healthcare – a strategy that aims to fit the right treatment to each patient in the best way possible.
Roche is the world’s largest biotech company, with truly differentiated medicines in oncology, immunology, infectious diseases, ophthalmology and diseases of the central nervous system. Roche is also the world leader in in vitro diagnostics and tissue-based cancer diagnostics, and a frontrunner in diabetes management.
Founded in 1896, Roche continues to search for better ways to prevent, diagnose and treat diseases and make a sustainable contribution to society. The company also aims to improve patient access to medical innovations by working with all relevant stakeholders. More than thirty medicines developed by Roche are included in the World Health Organization Model Lists of Essential Medicines, among them life-saving antibiotics, antimalarials and cancer medicines. Moreover, for the twelfth consecutive year, Roche has been recognised as one of the most sustainable companies in the Pharmaceuticals Industry by the Dow Jones Sustainability Indices (DJSI).
The Roche Group, headquartered in Basel, Switzerland, is active in over 100 countries and in 2020 employed more than 100,000 people worldwide. In 2020, Roche invested CHF 12.2 billion in R&D and posted sales of CHF 58.3 billion. Genentech, in the United States, is a wholly owned member of the Roche Group. Roche is the majority shareholder in Chugai Pharmaceutical, Japan
Final analysis from the phase IIIb STASEY study, including data from 193 people with haemophilia A, further support the benefit/risk profile of Hemlibra, with no new safety signals identified
STASEY is one of the largest open-label studies primarily assessing safety and tolerability of a medicine for people with haemophilia A with factor VIII inhibitors
Hemlibra also continued to demonstrate effective bleed control with a high proportion of participants (82.6%) achieving zero treated bleeds
Basel, 19 July 2021 - Roche (SIX: RO, ROG; OTCQX: RHHBY) today announced results from the final analysis of the phase IIIb STASEY study, which confirm the favourable safety profile of Hemlibra® (emicizumab), consistent with the phase III HAVEN clinical programme.1,2,3,4 In the analysis, no new safety signals were identified with longer-term Hemlibra treatment in adults and adolescents with haemophilia A with inhibitors to factor VIII, the clotting protein that is missing or defective in people with haemophilia A. The data were presented at the virtual International Society on Thrombosis and Haemostasis (ISTH) 2021 Congress, 17-21 July 2021.
“As the treatment landscape evolves, determining the long-term benefit/risk profile of medicines for people living with haemophilia A remains a top priority for the community,” said Levi Garraway, M.D., Ph.D., Roche’s Chief Medical Officer and Head of Global Product Development. “These results provide further confidence in Hemlibra’s favourable safety profile in people with haemophilia A with factor VIII inhibitors, who have historically faced significant treatment challenges.”
Nearly one in three people with haemophilia A develop factor VIII inhibitors, antibodies that bind to and block the efficacy of replacement factor VIII.5 People with haemophilia A with inhibitors are at greater risk of more frequent bleeding, including life-threatening bleeds, and may face greater challenges in their day-to-day lives than people with haemophilia A who do not have inhibitors.6 Hemlibra has been approved in more than 100 countries worldwide for the treatment of people with haemophilia A with factor VIII inhibitors.
The final analysis of the STASEY study included data from 193 people with haemophilia A with factor VIII inhibitors, who received Hemlibra prophylaxis once-weekly for up to two years (median treatment duration of 103.1 weeks).1 The analysis did not show any new cases of thrombotic microangiopathy or serious thrombotic events (adverse events [AEs] that have been observed in people with bleeding disorders) related to Hemlibra.1 The most common AEs occurring in 10% or more of people in the STASEY study were joint pain (arthralgia; 17.1%), common cold symptoms (nasopharyngitis; 15.5%), headache (15.0%), injection site reaction (ISR; 11.4%) and fever (pyrexia; 10.9%). Thirty-five (18.1%) people reported a Hemlibra-related AE, with ISRs being the most common (9.8%).1
In addition, the STASEY study reinforced that Hemlibra is associated with a low incidence of anti-drug antibody (ADA) development. Ten participants (5.2%) tested positive for ADAs, five (2.6%) of whom were classified as having ADAs that were neutralising in vitro.1 In all ten participants, ADA development did not affect the efficacy or safety of Hemlibra; none of the participants had ADAs that resulted in a decrease in Hemlibra plasma concentration, and none of the ADA-positive participants experienced a treated bleed. In addition, the ADAs disappeared over time, as all study participants tested negative for ADAs at their last visit.1
Hemlibra also continued to demonstrate effective bleed control in the STASEY study, with 82.6% of participants experiencing no bleeding episodes that required treatment. Annualised bleeding rates were consistent with previously reported observations from the pivotal HAVEN studies.1,2,3,4
Hemlibra is approved to treat people with haemophilia A with factor VIII inhibitors in more than 100 countries worldwide and people with haemophilia A without factor VIII inhibitors in more than 80 countries worldwide, including the US, EU and Japan. Hemlibra has been studied in one of the largest clinical trial programmes in haemophilia A with and without factor VIII inhibitors, including eight phase III studies.
Hemlibra is a bispecific factor IXa- and factor X-directed antibody. It is designed to bring together factor IXa and factor X, proteins involved in the natural coagulation cascade, and restore the blood clotting process for people with haemophilia A. Hemlibra is a prophylactic (preventative) treatment that can be administered by an injection of a ready-to-use solution under the skin (subcutaneously) once-weekly, every two weeks or every four weeks (after an initial once-weekly dose for the first four weeks). Hemlibra was created by Chugai Pharmaceutical Co., Ltd. and is being co-developed globally by Chugai, Roche and Genentech. It is marketed in the United States by Genentech as Hemlibra (emicizumab-kxwh), with kxwh as the suffix designated in accordance with Nonproprietary Naming of Biological Products Guidance for Industry issued by the U.S. Food and Drug Administration.
Haemophilia A is an inherited, serious disorder in which a person’s blood does not clot properly, leading to uncontrolled and often spontaneous bleeding. Haemophilia A affects around 900,000 people worldwide,5,7 approximately 35-39% of whom have a severe form of the disorder.7 People with haemophilia A either lack or do not have enough of a clotting protein called factor VIII. In a healthy person, when a bleed occurs, factor VIII brings together the clotting factors IXa and X, which is a critical step in the formation of a blood clot to help stop bleeding. Depending on the severity of their disorder, people with haemophilia A can bleed frequently, especially into their joints or muscles.5 These bleeds can present a significant health concern as they often cause pain and can lead to chronic swelling, deformity, reduced mobility, and long-term joint damage.8 A serious complication of treatment is the development of inhibitors to factor VIII replacement therapies.9 Inhibitors are antibodies developed by the body’s immune system that bind to and block the efficacy of replacement factor VIII,10 making it difficult, if not impossible, to obtain a level of factor VIII sufficient to control bleeding.
Roche has been developing medicines for people with malignant and non-malignant blood diseases for over 20 years; our experience and knowledge in this therapeutic area runs deep. Today, we are investing more than ever in our effort to bring innovative treatment options to patients across a wide range of haematologic diseases. Our approved medicines include MabThera®/Rituxan® (rituximab), Gazyva®/Gazyvaro® (obinutuzumab), Polivy® (polatuzumab vedotin), Venclexta®/Venclyxto® (venetoclax) in collaboration with AbbVie, and Hemlibra® (emicizumab). Our pipeline of investigational haematology medicines includes T-cell engaging bispecific antibodies, glofitamab and mosunetuzumab, targeting both CD20 and CD3, and cevostamab, targeting FcRH5 and CD3; Tecentriq® (atezolizumab), a monoclonal antibody designed to bind with PD-L1; and crovalimab, an anti-C5 antibody engineered to optimise complement inhibition. Our scientific expertise, combined with the breadth of our portfolio and pipeline, also provides a unique opportunity to develop combination regimens that aim to improve the lives of patients even further.
Roche is a global pioneer in pharmaceuticals and diagnostics focused on advancing science to improve people’s lives. The combined strengths of pharmaceuticals and diagnostics under one roof have made Roche the leader in personalised healthcare – a strategy that aims to fit the right treatment to each patient in the best way possible.
Roche is the world’s largest biotech company, with truly differentiated medicines in oncology, immunology, infectious diseases, ophthalmology and diseases of the central nervous system. Roche is also the world leader in in vitro diagnostics and tissue-based cancer diagnostics, and a frontrunner in diabetes management.
Founded in 1896, Roche continues to search for better ways to prevent, diagnose and treat diseases and make a sustainable contribution to society. The company also aims to improve patient access to medical innovations by working with all relevant stakeholders. More than thirty medicines developed by Roche are included in the World Health Organization Model Lists of Essential Medicines, among them life-saving antibiotics, antimalarials and cancer medicines. Moreover, for the twelfth consecutive year, Roche has been recognised as one of the most sustainable companies in the Pharmaceuticals Industry by the Dow Jones Sustainability Indices (DJSI).
The Roche Group, headquartered in Basel, Switzerland, is active in over 100 countries and in 2020 employed more than 100,000 people worldwide. In 2020, Roche invested CHF 12.2 billion in R&D and posted sales of CHF 58.3 billion. Genentech, in the United States, is a wholly owned member of the Roche Group. Roche is the majority shareholder in Chugai Pharmaceutical, Japan
Roche will present the final analysis from the phase IIIb STASEY study, reinforcing the safety and efficacy profile of Hemlibra in a broad range of people with haemophilia A with factor VIII inhibitors1
Spark Therapeutics will share updated data from the ongoing phase I/II clinical trial of investigational gene therapy SPK-8011, demonstrating that hepatocyte expression of factor VIII can be stable and durable up to four years following vector administration, with an acceptable safety profile2
These data reinforce Roche's continued focus on advancing care for people living with haemophilia A and provide additional insights into the long-term efficacy and safety profile of Hemlibra, building on the results previously observed in the phase III HAVEN studies
Basel, 02 July 2021 - Roche (SIX: RO, ROG; OTCQX: RHHBY) today announced that new data from its haemophilia A clinical programme will be presented at the virtual International Society on Thrombosis and Haemostasis (ISTH) 2021 Congress, from 17-21 July 2021. Data will include the final analysis from the phase IIIb STASEY study of Hemlibra® (emicizumab) and updated data from the phase I/II study of SPK-8011, an AAV-based gene therapy in development by Spark Therapeutics (a member of the Roche Group).1,2
“We’re excited to present new data from our haemophilia A programme at the ISTH 2021 Congress,” said Levi Garraway, M.D., Ph.D., Roche’s Chief Medical Officer and Head of Global Product Development. “These data reinforce our continued commitment to developing transformational therapies for the haemophilia A community and advancing understanding of the long-term efficacy and safety profile of Hemlibra.”
Haemophilia A is a serious, inherited bleeding disorder in which a person’s blood doesn't clot properly, as they either lack or do not have enough of a clotting protein called factor VIII. This can lead to uncontrolled bleeding, either spontaneously or after minor trauma. These bleeds can present a significant health concern as they often cause pain and can lead to chronic swelling, deformity, reduced mobility, and long-term joint damage.3 The development of factor VIII inhibitors can be a significant challenge in the treatment of people with haemophilia A as they bind to and block the efficacy of replacement factor VIII.4
The STASEY study is one of the largest open-label studies primarily assessing the safety and tolerability of a medicine for adults and adolescents with haemophilia A with factor VIII inhibitors. Final data from the STASEY study, evaluating the safety and tolerability of Hemlibra prophylaxis in adults and adolescents with haemophilia A with factor VIII inhibitors, will be presented at the congress.1 These results confirm the favourable safety profile of Hemlibra, as previously demonstrated in the phase III HAVEN clinical trials.1,5,6,7
Spark Therapeutics will share updated data from the ongoing phase I/II clinical trial of SPK-8011, an investigational AAV-based gene therapy developed for the treatment of haemophilia A. These data demonstrate that hepatocyte expression of factor VIII can be stable and durable for up to four years following vector administration, with an acceptable safety profile.2
“We are looking forward to sharing data on our investigational gene therapy, SPK-8011, which is being evaluated in the largest phase I/II gene therapy trial in haemophilia A to date, and which reinforces Spark’s mission to bring a novel gene therapy option to persons with haemophilia A,” said Gallia Levy, M.D., Ph.D., Chief Medical Officer, Spark Therapeutics.
Updated results from Spark’s ongoing phase I/II study of investigational SPK-8011 will be shared as an oral presentation during the meeting. These data highlight the safety profile and durability of SPK-8011 in 18 participants, up to four years following vector administration with SPK-8011 in four dose cohorts, ranging from 5x1011 vg/kg to 2×1012 vg/kg, with results showing a 93% reduction in annualised bleed rate (ABR) and a 97% reduction in annualised infusion rate.
Final data from the STASEY study presented at the congress demonstrate that Hemlibra is effective, with ABRs consistent with observations reported from the pivotal HAVEN studies.1,5,6,7 Additionally, no new safety signals were identified in adults and adolescents with haemophilia A with factor VIII inhibitors, consistent with previous safety observations.1
Roche will also present a retrospective analysis comparing ABR and Hemlibra concentrations among obese and non-obese adults with haemophilia A from pooled data from the HAVEN 1, 3, and 4 studies.8 These data suggest that body weight does not significantly impact the efficacy of Hemlibra, regardless of dosing regimen, demonstrating that Hemlibra offers an effective, well tolerated treatment, with flexible dosing options, in obese and non-obese people with haemophilia A.
Additionally, an analysis of data from the 2017 CHESS PAEDs (Cost of Haemophilia across Europe: a Socioeconomic Survey in the Paediatric Population) study, examining the association between physical activity levels and bleed rates in children with haemophilia A, will be presented.9 Results from this analysis demonstrate the potential treatment needs and clinical burden in physically active children with moderate and severe haemophilia A receiving factor VIII replacement therapy.
Key abstracts from Roche and Spark that will be presented at ISTH can be found in the table below.
Follow Roche and Spark on Twitter via @Roche and @Spark_tx respectively, and keep up to date with ISTH 2021 Congress news and updates by using the hashtag #ISTH2021.
Hemlibra is approved for routine prophylaxis of bleeding episodes in people with haemophilia A with and without factor VIII inhibitors in over 100 countries worldwide for those with inhibitors and over 80 countries for those without inhibitors, in adults and children, ages newborn and older. Hemlibra is a bispecific factor IXa- and factor X-directed antibody. It is designed to bring together factor IXa and factor X, proteins involved in the natural coagulation cascade, and restore the blood clotting process for people with haemophilia A. Hemlibra is a prophylactic (preventative) treatment that can be administered by an injection of a ready-to-use solution under the skin (subcutaneously) once-weekly, every two weeks or every four weeks (after an initial once-weekly dose for the first four weeks). Hemlibra was created by Chugai Pharmaceutical Co., Ltd. and is being co-developed globally by Chugai, Roche and Genentech. It is marketed in the United States by Genentech as Hemlibra (emicizumab-kxwh), with kxwh as the suffix designated in accordance with Nonproprietary Naming of Biological Products Guidance for Industry issued by the US Food and Drug Administration.
Investigational SPK-8011, a novel bio-engineered adeno-associated viral (AAV) vector utilizing the AAV-LK03 capsid, also referred to as Spark200, contains a codon-optimized human factor VIII gene under the control of a liver-specific promoter. The Food and Drug Administration (FDA) granted orphan-disease designation and breakthrough therapy designation in the U.S., while the European Commission has granted orphan designation to SPK-8011.
Haemophilia A is an inherited, serious disorder in which a person’s blood does not clot properly, leading to uncontrolled and often spontaneous bleeding. Haemophilia A affects around 900,000 people worldwide,10,11 approximately 35-39% of whom have a severe form of the disorder.11 People with haemophilia A either lack or do not have enough of a clotting protein called factor VIII. In a healthy person, when a bleed occurs, factor VIII brings together the clotting factors IXa and X, which is a critical step in the formation of a blood clot to help stop bleeding. Depending on the severity of their disorder, people with haemophilia A can bleed frequently, especially into their joints or muscles.10 These bleeds can present a significant health concern as they often cause pain and can lead to chronic swelling, deformity, reduced mobility, and long-term joint damage.3 A serious complication of treatment is the development of inhibitors to factor VIII replacement therapies.12 Inhibitors are antibodies developed by the body’s immune system that bind to and block the efficacy of replacement factor VIII,13 making it difficult, if not impossible, to obtain a level of factor VIII sufficient to control bleeding.
We believe gene therapy has the potential to revolutionise medicine and improve the lives of patients with genetic and other serious diseases. Pairing Roche’s long-standing commitment to developing medicines in haemophilia with Spark Therapeutics’ proven gene therapy expertise brings together the best team of collaborators researching gene therapies in haemophilia A.
It is our aligned objective to develop gene therapies for haemophilia A that, with the lowest effective dose and the optimal immunomodulatory regimen, demonstrate safety, predictability, efficacy, and durability for patients.
At Spark Therapeutics, a fully integrated, commercial company committed to discovering, developing and delivering gene therapies, we challenge the inevitability of genetic diseases, including blindness, haemophilia, lysosomal storage disorders and neurodegenerative diseases. We currently have four programs in clinical trials. At Spark, a member of the Roche Group, we see the path to a world where no life is limited by genetic disease
Roche has been developing medicines for people with malignant and non-malignant blood diseases for over 20 years; our experience and knowledge in this therapeutic area runs deep. Today, we are investing more than ever in our effort to bring innovative treatment options to patients across a wide range of haematologic diseases. Our approved medicines include MabThera®/Rituxan® (rituximab), Gazyva®/Gazyvaro® (obinutuzumab), Polivy® (polatuzumab vedotin), Venclexta®/Venclyxto® (venetoclax) in collaboration with AbbVie, and Hemlibra® (emicizumab). Our pipeline of investigational haematology medicines includes T-cell engaging bispecific antibodies, glofitamab and mosunetuzumab, targeting both CD20 and CD3, and cevostamab, targeting FcRH5 and CD3; Tecentriq® (atezolizumab), a monoclonal antibody designed to bind with PD-L1; and crovalimab, an anti-C5 antibody engineered to optimise complement inhibition. Our scientific expertise, combined with the breadth of our portfolio and pipeline, also provides a unique opportunity to develop combination regimens that aim to improve the lives of patients even further.
Roche is a global pioneer in pharmaceuticals and diagnostics focused on advancing science to improve people’s lives. The combined strengths of pharmaceuticals and diagnostics under one roof have made Roche the leader in personalised healthcare – a strategy that aims to fit the right treatment to each patient in the best way possible.
Roche is the world’s largest biotech company, with truly differentiated medicines in oncology, immunology, infectious diseases, ophthalmology and diseases of the central nervous system. Roche is also the world leader in in vitro diagnostics and tissue-based cancer diagnostics, and a frontrunner in diabetes management.
Founded in 1896, Roche continues to search for better ways to prevent, diagnose and treat diseases and make a sustainable contribution to society. The company also aims to improve patient access to medical innovations by working with all relevant stakeholders. More than thirty medicines developed by Roche are included in the World Health Organization Model Lists of Essential Medicines, among them life-saving antibiotics, antimalarials and cancer medicines. Moreover, for the twelfth consecutive year, Roche has been recognised as one of the most sustainable companies in the Pharmaceuticals Industry by the Dow Jones Sustainability Indices (DJSI).
The Roche Group, headquartered in Basel, Switzerland, is active in over 100 countries and in 2020 employed more than 100,000 people worldwide. In 2020, Roche invested CHF 12.2 billion in R&D and posted sales of CHF 58.3 billion. Genentech, in the United States, is a wholly owned member of the Roche Group. Roche is the majority shareholder in Chugai Pharmaceutical, Japan
The authorisation enables emergency use of Actemra/RoActemra for the treatment of COVID-19 in hospitalised adult and paediatric patients
Basel, 25 June 2021 - Roche (SIX: RO, ROG; OTCQX: RHHBY) announced today that the U.S. Food and Drug Administration (FDA) has issued an Emergency Use Authorization (EUA) for intravenous Actemra/RoActemra® (tocilizumab) for the treatment of COVID-19 in hospitalised adults and paediatric patients (two years of age and older) who are receiving systemic corticosteroids and require supplemental oxygen, non-invasive or invasive mechanical ventilation, or extracorporeal membrane oxygenation (ECMO). The EUA is based on results from four randomised, controlled studies that evaluated Actemra/RoActemra for the treatment of COVID-19 in more than 5,500 hospitalised patients. The results of these studies suggest that Actemra/RoActemra may improve outcomes in patients receiving corticosteroids and requiring supplemental oxygen or breathing support.
“Even with the availability of vaccines and declines in deaths from COVID-19 in various parts of the world, we continue to see new hospitalisations from severe forms of the disease,” said Levi Garraway, M.D., Ph.D., Roche’s Chief Medical Officer and Head of Global Product Development. “We are pleased that Actemra/RoActemra is now authorised as an option that may help improve outcomes for adults and children hospitalised with COVID-19 in the United States.”
The four randomised, controlled studies included in the EUA submission investigated the safety and efficacy of Actemra/RoActemra in more than 5,500 hospitalised patients with COVID-19. The RECOVERY Actemra/RoActemra study was led by researchers in the United Kingdom and included more than 4,000 hospitalised COVID-19 patients. Roche-sponsored global trials included the placebo-controlled EMPACTA, COVACTA and REMDACTA studies. There have been no new safety signals identified for Actemra/RoActemra in any of these studies. The most common adverse reactions seen (incidence ≥ 3%) are constipation, anxiety, diarrhoea, insomnia, hypertension and nausea.
The U.S. FDA Letter of Authorisation and Fact Sheets for patients and health care professionals are available for download with the latest information on this EUA. For more information on how Roche is responding to the global COVID-19 pandemic, please visit our COVID-19 response page.
In these exceptional times, Roche stands together with society, governments, healthcare providers and all those working to overcome the pandemic.
Actemra/RoActemra has not been approved by the U.S. FDA in this setting, but the U.S. FDA has made Actemra/RoActemra available under an emergency access mechanism called an EUA as a treatment for certain patients with COVID-19. There is limited information known about the safety or effectiveness of using Actemra/RoActemra to treat people in the hospital with COVID-19. The EUA is supported by a Secretary of Health and Human Services (HHS) declaration that circumstances exist to justify the emergency use of drugs and biological products during the COVID-19 pandemic. The authorisation is temporary and does not replace the formal review and approval process. Actemra/RoActemra is authorised under the EUA only for the duration of the declaration that circumstances exist justifying the authorisation of the emergency use of Actemra/RoActemra under Section 564(b)(1) of the Act, 21 U.S.C.§ 360bbb-3(b)(1), unless the authorisation is terminated or revoked sooner. Roche has existing distribution channels established to ship Actemra/RoActemra to hospitals across the United States.
Roche’s clinical trial programme evaluated the safety and efficacy of Actemra/RoActemra in hospitalised patients with COVID-19. Actemra/RoActemra is not approved for this use in any country and there is limited information known about the safety or effectiveness of using Actemra to treat people in the hospital with COVID-19. COVACTA and EMPACTA were the first two global phase III, multicentre, randomised, placebo-controlled studies of Actemra/RoActemra in patients hospitalised with COVID-19 associated pneumonia. COVACTA was conducted in collaboration with the Biomedical Advanced Research and Development Authority (BARDA), part of the Office of the Assistant Secretary for Preparedness and Response at the United States Department of Health and Human Services (HHS). EMPACTA aimed to address research questions about the safety and efficacy of Actemra in underserved populations by emphasising enrollment from minority patients often underrepresented in clinical trials. Both studies were published in the New England Journal of Medicine. Roche also partnered with Gilead Sciences, Inc., on REMDACTA, a phase III, randomised, double-blind, multicentre study to evaluate the safety and efficacy of Actemra/RoActemra plus Veklury® (remdesivir), versus placebo plus Veklury, in hospitalised patients with severe COVID-19 associated pneumonia.
Actemra/RoActemra was the first humanised interleukin-6 (IL-6) receptor antagonist approved for the treatment of adult patients with moderately to severely active rheumatoid arthritis (RA) who have used one or more disease-modifying antirheumatic drugs (DMARDs), such as methotrexate (MTX), that did not provide enough relief. The extensive Actemra/RoActemra RA IV clinical development programme included five phase III clinical studies and enrolled more than 4,000 people with RA in 41 countries. The Actemra/RoActemra RA subcutaneous clinical development programme included two phase III clinical studies and enrolled more than 1,800 people with RA in 33 countries. Actemra/RoActemra subcutaneous injection is also approved for the treatment of adult patients with giant cell arteritis (GCA), for the treatment of patients two years of age and older with active polyarticular juvenile idiopathic arthritis (PJIA) or active systemic juvenile idiopathic arthritis (SJIA), and for slowing the rate of decline in pulmonary function in adult patients with systemic sclerosis-associated interstitial lung disease (SSc-ILD). In addition, Actemra/RoActemra is also approved in the IV formulation for patients two years of age and older with active PJIA, SJIA or CAR T cell-induced cytokine release syndrome (CRS). Actemra/RoActemra is not approved for subcutaneous use in people with CRS. It is not known if Actemra is safe and effective in children with PJIA, SJIA or CRS under two years of age or in children with conditions other than PJIA, SJIA or CRS. Actemra is intended for use under the guidance of a healthcare practitioner.
As a leading healthcare company we are doing all we can to support countries in their fight against COVID-19 and minimising its impact. We have developed a growing number of diagnostic solutions that help to detect and diagnose the infection, as well as providing digital support to healthcare systems. We also continue to identify, develop and support potential therapies which can play a role in treating the disease.
The impact of COVID-19 goes beyond those who contract it. That is why we are working with healthcare providers, laboratories, authorities and organisations to help make sure patients continue to receive the tests, treatment and care they need during these challenging times. Building on a longstanding tradition of partnerships, we are working together with governments and others to make healthcare stronger and more sustainable in the future.
Reliable, high-quality testing is essential to help healthcare systems overcome this pandemic and Roche has so far launched 16 diagnostics solutions to help minimise the impact of COVID-19. As soon as the novel SARS-CoV-2 virus was sequenced in early 2020, we got to work. On 13 March 2020 we became the first company to receive U.S. Food and Drug Administration (FDA) Emergency Use Authorization (EUA) for a high-volume molecular test to detect the virus. Since then, we have continued to add a range of diagnostics solutions to our global portfolio to help in the fight against COVID-19. In addition to the gold standard PCR test, we have developed antigen tests to help diagnose the virus in settings where there is limited molecular laboratory infrastructure, rapid antigen where the virus can be detected on the spot, tests that can test for both flu and COVID-19 at the same time, both high throughput and at the point of care, and tests that can detect virus antibodies that can help monitor the spread of the virus and can also support in vaccine development. On 16 March 2021 the SARS-CoV-2 variant test was launched, designed to detect key spike mutations.
Aside from these tests we have also looked at how we can support care for patients who have COVID-19, receiving an U.S. FDA EUA for the Elecsys® IL-6 test to assist in identifying severe inflammatory response in patients with confirmed COVID-19, as well as launching Roche v-TAC, a digital algorithm that could help simplify the screening, diagnosis and monitoring of respiratory-compromised patients with COVID-19. Roche is working closely with governments and health authorities around the world, and has significantly increased production to support availability of tests globally.
Roche is actively involved in understanding the potential of the existing portfolio and is researching options for the future. In 2020, Roche entered into a number of new partnerships, including with Gilead, Regeneron and Atea, to develop, manufacture and distribute molecules that can potentially both treat and prevent COVID-19.
In October, Roche announced a partnership with Atea Pharmaceuticals to jointly develop the investigational compound AT-527. If approved, Atea will distribute AT-527 in the United States (US) and Roche will be responsible for global manufacturing and distribution outside the US. Atea’s compound has the potential to be the first oral antiviral to treat COVID-19 patients outside the hospital setting as well as in the hospital. Its anticipated formulation (pill) may help to facilitate access to a broad patient population.
In November, our partner Regeneron received U.S. FDA EUA for casirivimab and imdevimab, its investigational antiviral antibody combination, for the treatment of recently diagnosed patients with mild to moderate COVID-19 who are at high risk of progressing to severe COVID-19 and/or hospitalisation. The antibody cocktail has been studied in two phase I-III adaptive clinical trials for the treatment of COVID-19 and in a phase III trial for the prevention of the disease. As part of the global partnership with Regeneron, we are committing a significant amount of manufacturing capacity and are working to expand supply of this antibody combination beyond the US to as many people as possible.
In addition, we are exploring the potential of our investigational molecules and existing portfolio: For example, Roche has initiated three global phase III clinical trials investigating the safety and efficacy of Actemra/RoActemra in COVID-19 associated pneumonia (COVACTA, EMPACTA and REMDACTA). Roche will continue to monitor the evolving clinical evidence for Actemra/RoActemra in this setting.
Roche is a global pioneer in pharmaceuticals and diagnostics focused on advancing science to improve people’s lives. The combined strengths of pharmaceuticals and diagnostics under one roof have made Roche the leader in personalised healthcare – a strategy that aims to fit the right treatment to each patient in the best way possible.
Roche is the world’s largest biotech company, with truly differentiated medicines in oncology, immunology, infectious diseases, ophthalmology and diseases of the central nervous system. Roche is also the world leader in in vitro diagnostics and tissue-based cancer diagnostics, and a frontrunner in diabetes management.
Founded in 1896, Roche continues to search for better ways to prevent, diagnose and treat diseases and make a sustainable contribution to society. The company also aims to improve patient access to medical innovations by working with all relevant stakeholders. More than thirty medicines developed by Roche are included in the World Health Organization Model Lists of Essential Medicines, among them life-saving antibiotics, antimalarials and cancer medicines. Moreover, for the twelfth consecutive year, Roche has been recognised as one of the most sustainable companies in the Pharmaceuticals Industry by the Dow Jones Sustainability Indices (DJSI).
The Roche Group, headquartered in Basel, Switzerland, is active in over 100 countries and in 2020 employed more than 100,000 people worldwide. In 2020, Roche invested CHF 12.2 billion in R&D and posted sales of CHF 58.3 billion. Genentech, in the United States, is a wholly owned member of the Roche Group. Roche is the majority shareholder in Chugai Pharmaceutical, Japan
Data for EVRYSDI reinforce safety profile and efficacy in a broad spinal muscular atrophy (SMA) population, following recent EU approval
Data for ENSPRYNG in neuromyelitis optica spectrum disorder (NMOSD) build on safety profile and efficacy following recent CHMP opinion, including in adults with concomitant autoimmune diseases (CAIDs)
OCREVUS data continue to show consistent benefit on slowing disease progression in relapsing MS (RMS) and primary progressive MS (PPMS)
Additional presentations in Alzheimer’s disease (AD), Huntington’s disease (HD) and Parkinson’s disease (PD) continue to contribute to understanding of these complex neurological disorders
Basel, 15 June 2021 - Roche (SIX: RO, ROG; OTCQX: RHHBY) today announced that data across its growing neuroscience portfolio will be presented at the 7th Congress of the European Academy of Neurology (EAN) Annual Meeting being held virtually 19-22 June, 2021. These new and encore data demonstrate Roche’s commitment to advancing the clinical understanding of a broad range of neurological disorders with the goal of meeting the needs of people living with both the rarest and most common conditions.“
Our data at EAN and recent European regulatory milestones for EVRYSDI and ENSPRYNG reflect our continued commitment to discovering and developing breakthrough medicines for challenging neurological conditions.” said Levi Garraway, M.D., Ph.D., Roche’s Chief Medical Officer and Head of Global Product Development. “We are honoured to work with our partners and the broader community to accelerate progress across our neuroscience portfolio with the goal of transforming the lives of many people living with neurological disorders.”
Roche will present updated data from across the extensive EVRYSDI™ (risdiplam) clinical development programme, designed to represent a broad spectrum of people living with SMA, including those who have previously been treated with another SMA medication.
Among the five abstracts featured, new data includes 12-month safety, pharmacodynamic and interim exploratory efficacy data from the JEWELFISH study of EVRYSDI in people previously treated with SMA-targeting therapies across a broad range of ages (1–60 years), SMA types (1–3) and SMN2 copy number (1-5).
Data from SUNFISH Part 2 supporting the safety profile and efficacy of EVRYSDI in people aged 2-25 years with SMA Types 2 or non-ambulant Type 3 after two years of treatment will also be shared, as well as updated 2-year safety and efficacy data from the pivotal FIREFISH Parts 1 and 2 showing continued improvements in survival and motor milestones in infants aged one to seven months with Type 1 SMA.
In addition, preliminary safety and efficacy data from the RAINBOWFISH study of EVRYSDI treatment in pre-symptomatic babies from birth to six weeks will be presented, along with longer-term safety data from a separate pooled analysis of the FIREFISH, SUNFISH, JEWELFISH and RAINBOWFISH trials.
EVRYSDI, the first and only at home SMA treatment, is now approved in 42 countries, including the U.S. and EU. More than 3,000 patients have been treated with EVRYSDI in clinical trials, compassionate use and real-world settings.
Roche will share three sets of data on people living with NMOSD, including an analysis of the Phase III SAkuraStar and SAkuraSky clinical trials which show ENSPRYNG® (satralizumab) significantly reduced risk of relapse vs placebo in adults with AQP4-IgG seropositive (AQP4-IgG+) NMOSD, with a favourable safety profile.
A separate pooled data analysis from the SAkura studies will also be presented, reinforcing favourable safety and efficacy of ENSPRYNG in people with NMOSD, including those with concomitant autoimmune diseases (CAIDs). Results from a new claims-based algorithm to identify people with NMOSD and NMOSD relapses in German claims data will also be shared.
ENSPRYNG is currently approved in 24 countries and has received a positive CHMP opinion as the first and only subcutaneous treatment for adults and adolescents with AQP4-IgG seropositive NMOSD in the EU, enabling home dosing by a person with NMOSD or their care partner.
Four OCREVUS® (ocrelizumab) presentations will be shared, including an analysis of the Phase IIIb CASTING study in people with relapsing-remitting MS (RRMS) who had a suboptimal response to one or two prior disease-modifying therapies (DMTs). OCREVUS improved work productivity over two years, which correlated with reduced symptom burden and improvement in the physical and psychological impacts of MS.
Data from a separate subgroup analysis of the CASTING study will also be presented, continuing to show a favourable benefit-risk profile for OCREVUS and comparable safety outcomes regardless of age, type, or number of prior DMTs.
In addition, data from the OPERA and ORATORIO Phase III studies and their open-label extensions will be shared. A pooled analysis of the OPERA I and II studies, showed that OCREVUS reduced cerebellar atrophy in relapsing MS (RMS), compared with interferon beta-1a (IFN). An analysis of the open-label extension studies demonstrated that individuals initially treated with OCREVUS maintained lower cerebellar volume loss relative to baseline in both RMS and primary progressive MS (PPMS) during the open-label extension periods vs. those initially treated with comparators. OCREVUS is the first and only therapy approved for both RMS and PPMS with twice-yearly dosing, with over 200,000 patients treated globally.
Finally, a late-breaking oral abstract on risk factors for developing symptomatic or serious COVID-19 in patients treated with OCREVUS will be presented.
Roche will present an overview of the CareRing initiative, which engaged AD caregivers to input and provide guidance on trial design and protocol for the Phase IB/IIA Brain Shuttle AD trial, evaluating RG6102 in people with prodromal or mild-to-moderate AD.
RG6102 is a bispecific 2+1 monoclonal antibody that combines investigational gantenerumab with Roche’s Brain Shuttle technology. It is designed to be transported across the blood-brain barrier by engaging the transferrin receptor in order to achieve superior target engagement in the brain.
The protocol for a multinational qualitative study sponsored by Roche examining the burden of HD from a multidimensional, generational perspective (SEEING-HD) will be presented. Findings from this study will make an important contribution to advancing evidence-based care to improve the lives of individuals and families affected by HD.
Roche will share three presentations including further results from the Phase II PASADENA study evaluating the safety and efficacy of prasinezumab in early PD. In the PASADENA study, prasinezumab was the first anti-alpha-synuclein antibody to show evidence of slowing clinical decline of PD motor signs and demonstrated a favourable safety profile. These findings warrant further investigations in the recently initiated PADOVA study, studying the effect of prasinezumab in people with early Parkinson's disease receiving symptomatic therapy. In addition, data from two studies on digital health monitoring in PD will also be presented.
The full range of data from Roche’s clinical development programme in neuroscience being presented at EAN include:
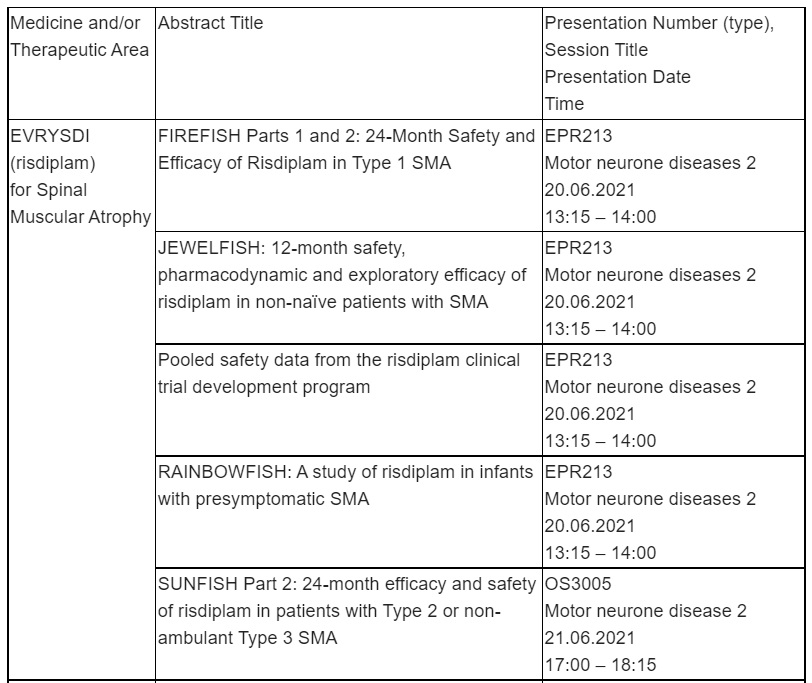


About EVRYSDI™ (risdiplam)
EVRYSDI is a survival of motor neuron 2 (SMN2) splicing modifier designed to treat SMA by increasing production of the survival of motor neuron (SMN) protein. SMN protein is found throughout the body and is critical for maintaining healthy motor neurons and movement. EVRYSDI is administered daily at home in liquid form by mouth or by feeding tube.
The U.S. Food and Drug Administration (FDA) and European Medicines Agency (EMA) approved EVRYSDI for the treatment of SMA in adults and children 2 months of age and older. EVRYSDI was granted PRIME designation by the EMA in 2018 and Orphan Drug Designation by FDA and EMA in 2017 and 2019, respectively. At this time, EVRYSDI has been approved in seven countries and submitted in 57, including the EU 27 and Norway and Iceland.
ENSPRYNG, which was designed by Chugai, a member of the Roche Group, is a humanised monoclonal antibody that targets interleukin-6 (IL-6) receptor activity. The cytokine IL-6 is believed to be a key driver in NMOSD, triggering the inflammation cascade and leading to damage and disability. ENSPRYNG was designed using novel recycling antibody technology which, compared to conventional technology, allows for longer duration of the antibody and subcutaneous dosing every four weeks.
Positive Phase III results for ENSPRYNG, as both monotherapy and in combination with baseline immunosuppressive therapy, suggest that IL-6 inhibition is an effective therapeutic approach for NMOSD. The Phase III clinical development programme for ENSPRYNG included two studies: SAkuraStar and SAkuraSky.
ENSPRYNG is currently approved in 24 countries, including the United States, Canada, Japan, China and Switzerland.
ENSPRYNG has been designated as an orphan drug in the U.S., Europe and Japan. In addition, it was granted Breakthrough Therapy Designation for the treatment of NMOSD by the FDA in December 2018.
OCREVUS is the first and only therapy approved for both RMS (including clinically isolated syndrome, RRMS and active, or relapsing, SPMS) and PPMS, with dosing every six months. OCREVUS is a humanised monoclonal antibody designed to target CD20-positive B cells, a specific type of immune cell thought to be a key contributor to myelin (nerve cell insulation and support) and axonal (nerve cell) damage. This nerve cell damage can lead to disability in people with MS. Based on preclinical studies, OCREVUS binds to CD20 cell surface proteins expressed on certain B cells, but not on stem cells or plasma cells, suggesting that important functions of the immune system may be preserved.
OCREVUS is administered by intravenous infusion every six months. The initial dose is given as two 300 mg infusions given two weeks apart. Subsequent doses are given as single 600 mg infusions.
Neuroscience is a major focus of research and development at Roche. Our goal is to pursue groundbreaking science to develop new treatments that help improve the lives of people with chronic and potentially devastating diseases.
Roche is investigating more than a dozen medicines for neurological disorders, including spinal muscular atrophy, multiple sclerosis, neuromyelitis optica spectrum disorder, Alzheimer’s disease, Huntington’s disease, Parkinson’s disease, Duchenne muscular dystrophy and autism spectrum disorder. Together with our partners, we are committed to pushing the boundaries of scientific understanding to solve some of the most difficult challenges in neuroscience today.
Roche is a global pioneer in pharmaceuticals and diagnostics focused on advancing science to improve people’s lives. The combined strengths of pharmaceuticals and diagnostics under one roof have made Roche the leader in personalised healthcare – a strategy that aims to fit the right treatment to each patient in the best way possible.
Roche is the world’s largest biotech company, with truly differentiated medicines in oncology, immunology, infectious diseases, ophthalmology and diseases of the central nervous system. Roche is also the world leader in in vitro diagnostics and tissue-based cancer diagnostics, and a frontrunner in diabetes management.
Founded in 1896, Roche continues to search for better ways to prevent, diagnose and treat diseases and make a sustainable contribution to society. The company also aims to improve patient access to medical innovations by working with all relevant stakeholders. More than thirty medicines developed by Roche are included in the World Health Organization Model Lists of Essential Medicines, among them life-saving antibiotics, antimalarials and cancer medicines. Moreover, for the twelfth consecutive year, Roche has been recognised as one of the most sustainable companies in the Pharmaceuticals Industry by the Dow Jones Sustainability Indices (DJSI).
The Roche Group, headquartered in Basel, Switzerland, is active in over 100 countries and in 2020 employed more than 100,000 people worldwide. In 2020, Roche invested CHF 12.2 billion in R&D and posted sales of CHF 58.3 billion. Genentech, in the United States, is a wholly owned member of the Roche Group. Roche is the majority shareholder in Chugai Pharmaceutical, Japan
Data for investigational CD20xCD3 bispecific antibodies and new combination regimens with Polivy showed enhanced clinical benefits for people with non-Hodgkin lymphoma in early studies
Basel, 4 June 2021 ‒ Roche (SIX: RO, ROG; OTCQX: RHHBY) today announced that new data on its investigational CD20xCD3 T-cell engaging bispecific antibodies, mosunetuzumab and glofitamab, and its first-in-class anti-CD79b antibody-drug conjugate, Polivy® (polatuzumab vedotin), in non-Hodgkin lymphoma (NHL) will be presented at the 2021 ASCO Annual Meeting from 4-8 June 2021.
“People with difficult-to-treat blood cancers such as non-Hodgkin lymphoma still need more options to help improve outcomes,” said Levi Garraway, M.D., Ph.D., Roche’s Chief Medical Officer and Head of Global Product Development. “We are encouraged by promising data from our emerging T-cell engaging bispecific antibodies, mosunetuzumab and glofitamab, and the antibody-drug conjugate, Polivy, that demonstrate the potential of these novel immunotherapeutic approaches for various groups of patients.”
While approximately 500,000 people worldwide are diagnosed with NHL each year, treatment options are currently limited and resistance to existing therapies or relapse following treatment is common.1 The most prevalent form of NHL, accounting for about 40% of newly diagnosed NHL cases, is an aggressive form called diffuse large B-cell lymphoma (DLBCL), that comes with a life expectancy of weeks or months if left untreated.2,3
In clinical trials to date, the investigational CD20xCD3 T-cell engaging bispecific antibodies, mosunetuzumab and glofitamab, have shown promising responses across multiple types of NHL, including relapsed or refractory (R/R) DLBCL and follicular lymphoma (FL). Pivotal data for these medicines are expected this year and Roche is targeting a regulatory filing for mosunetuzumab in FL by the end of 2021, following its U.S. Food and Drug Administration Breakthrough Therapy Designation granted in June 2020. Key data on mosunetuzumab and glofitamab to be presented at the meeting include:
Phase I NP30179 study investigating step-up dosing of glofitamab in heavily pre-treated R/R NHL, showed high, ongoing complete responses (CRs) and an acceptable safety profile. After a median follow-up of 6.3 months, results showed that glofitamab achieved a complete metabolic response rate, defined as the disappearance of metabolic tumour activity, of 71.4% in patients with aggressive (fast-growing) NHL. The most common adverse events (AEs) were cytokine release syndrome (CRS) (63.5%), neutropenia (38.5%), and pyrexia (32.7%); CRS events were mostly low grade and confined to the first cycle of treatment.4
Phase I/II GO40516 study of mosunetuzumab in combination with Polivy in R/R NHL showed promising efficacy and an acceptable safety profile. The regimen achieved a CR of 54.5% in all patients. Eighty six percent of patients evaluated had aggressive NHL, and these patients achieved a CR rate of 47.4%. The most frequent treatment-related AEs were neutropenia (45.4%), fatigue, nausea, and diarrhoea (all 36.4%) and CRS (9.1%; all Grade 1).5
Broad development programmes are ongoing for mosunetuzumab and glofitamab, including the phase III GO42909 trial investigating mosunetuzumab plus lenalidomide versus MabThera®/Rituxan® (rituximab) plus lenalidomide in R/R FL, which will soon be enrolling patients. For glofitamab, the phase III GO41944 open-label, randomised trial designed to evaluate the safety and efficacy of glofitamab plus gemcitabine and oxaliplatin (glofit-GemOx) versus MabThera/Rituxan plus GemOx in patients with R/R DLBCL, is also ongoing.
Roche is committed to pursuing treatment solutions that can be tailored to meet the various needs of both people living with NHL and healthcare professionals. Polivy is already a treatment option for people with R/R DLBCL and continues to show potential in multiple combinations. Key data at the meeting include:
New triplet combination of Polivy with MabThera/Rituxan and lenalidomide, which demonstrated promising activity in difficult-to-treat R/R DLBCL, based on results from the phase Ib/II GO29834 study. With a median follow-up of 9.7 months, median overall survival was 10.9 months (95% CI: 7.4–NE) and median progression-free survival was 6.3 months (95% CI: 4.5–9.7) in patients treated with the triplet. The most commonly reported Grade 3-4 AEs were neutropenia (58.0%), thrombocytopenia (14.0%), infections (14.0%) and anaemia (11.0%).6
As a leader in haematology development, Roche will continue to follow the science to expand and improve upon treatment options for healthcare providers and people with difficult-to-treat blood cancers.
Roche is currently developing two T-cell engaging bispecific antibodies, mosunetuzumab and glofitamab, designed to target CD20 on the surface of B-cells and CD3 on the surface of T-cells. This dual targeting activates and redirects a patient’s existing T-cells to engage and eliminate target B-cells by releasing cytotoxic proteins into the B-cells. Mosunetuzumab and glofitamab differ in their structures, and both are being developed by Roche as part of our ongoing strategy to explore multiple bispecific formats, to identify those that maximise potential clinical benefits for patients. Mosunetuzumab has a structure similar to that of a natural human antibody in that it has two ‘Fab’ regions, but is different from naturally-occurring antibodies in that one ‘Fab’ region targets CD20 and the other ‘Fab’ region targets CD3. Glofitamab is based on a novel structural format called ‘2:1’, which refers to the structure of the antibody. It is engineered to have two ‘Fab’ regions which bind to CD20, and one ‘Fab’ region which binds to CD3. The clinical development programmes for mosunetuzumab and glofitamab include ongoing investigations of these molecules as monotherapies and in combination with other medicines, for the treatment of people with CD20-positive B-cell non-Hodgkin lymphomas (NHL), including diffuse large B-cell lymphoma (DLBCL) and follicular lymphoma (FL).
Polivy is a first-in-class anti-CD79b antibody-drug conjugate (ADC). The CD79b protein is expressed specifically in the majority of B-cells, an immune cell impacted in some types of NHL, making it a promising target for the development of new therapies.7,8 Polivy binds to CD79b and destroys these B-cells through the delivery of an anti-cancer agent, which is thought to minimise the effects on normal cells.9,10 Polivy is being developed by Roche using Seagen ADC technology and is currently being investigated for the treatment of several types of NHL. Polivy is marketed in the US by Genentech as Polivy (polatuzumab vedotin-piiq), with piiq as the suffix designated in accordance with Nonproprietary Naming of Biological Products Guidance for Industry issued by the U.S. Food and Drug Administration.
The NP30179 study [NCT03075696] is a phase I/Ib, multicentre, open-label, dose-escalation study, evaluating the efficacy, safety, tolerability and pharmacokinetics of glofitamab. In this study, glofitamab is assessed as a single-agent and in combination with Gazyva®/Gazyvaro® (obinutuzumab), following pre-treatment with a one-time, fixed-dose of Gazyva/Gazyvaro, in people with relapsed or refractory (R/R) B-cell NHL. Outcome measures include overall response rate, complete response rate per Lugano 2014 criteria, maximum tolerated dose and tolerability.
The GO40516 study [NCT03671018] is a phase I/II, multicentre, open-label study, evaluating the efficacy, safety, tolerability and pharmacokinetics of mosunetuzumab in combination with Polivy in people with B-cell NHL. It consists of a dose finding portion followed by an expansion phase for second line or later (2L+) participants with R/R DLBCL and 2L+ R/R FL. Outcome measures include best overall response rate, maximum tolerated dose and tolerability.
The GO29834 study [NCT02600897] is a phase Ib/II, multicentre, open-label study, evaluating the efficacy and safety of Polivy with MabThera® /Rituxan® (rituximab) and lenalidomide in R/R DLBCL. Outcome measures include complete response and tolerability.
Roche has been developing medicines for people with malignant and non-malignant blood diseases for over 20 years; our experience and knowledge in this therapeutic area runs deep. Today, we are investing more than ever in our effort to bring innovative treatment options to patients across a wide range of haematologic diseases. Our approved medicines include MabThera/Rituxan, Gazyva/Gazyvaro, Polivy, Venclexta®/Venclyxto® (venetoclax) in collaboration with AbbVie, and Hemlibra® (emicizumab). Our pipeline of investigational haematology medicines includes T-cell engaging bispecific antibodies, glofitamab and mosunetuzumab, targeting both CD20 and CD3, and cevostamab, targeting FcRH5 and CD3; Tecentriq® (atezolizumab), a monoclonal antibody designed to bind with PD-L1; and crovalimab, an anti-C5 antibody engineered to optimise complement inhibition. Our scientific expertise, combined with the breadth of our portfolio and pipeline, also provides a unique opportunity to develop combination regimens that aim to improve the lives of patients even further.
Roche is a global pioneer in pharmaceuticals and diagnostics focused on advancing science to improve people’s lives. The combined strengths of pharmaceuticals and diagnostics under one roof have made Roche the leader in personalised healthcare – a strategy that aims to fit the right treatment to each patient in the best way possible.
Roche is the world’s largest biotech company, with truly differentiated medicines in oncology, immunology, infectious diseases, ophthalmology and diseases of the central nervous system. Roche is also the world leader in in vitro diagnostics and tissue-based cancer diagnostics, and a frontrunner in diabetes management.
Founded in 1896, Roche continues to search for better ways to prevent, diagnose and treat diseases and make a sustainable contribution to society. The company also aims to improve patient access to medical innovations by working with all relevant stakeholders. More than thirty medicines developed by Roche are included in the World Health Organization Model Lists of Essential Medicines, among them life-saving antibiotics, antimalarials and cancer medicines. Moreover, for the twelfth consecutive year, Roche has been recognised as one of the most sustainable companies in the Pharmaceuticals Industry by the Dow Jones Sustainability Indices (DJSI).
The Roche Group, headquartered in Basel, Switzerland, is active in over 100 countries and in 2020 employed more than 100,000 people worldwide. In 2020, Roche invested CHF 12.2 billion in R&D and posted sales of CHF 58.3 billion. Genentech, in the United States, is a wholly owned member of the Roche Group. Roche is the majority shareholder in Chugai Pharmaceutical, Japan