If approved, Tecentriq subcutaneous (SC) would be the first injectable PD-(L)1 cancer immunotherapy in the EU, cutting treatment time by approx. 80%1
The CHMP recommended Tecentriq SC for all indications of intravenous (IV) Tecentriq, including certain types of lung, liver, bladder and breast cancer2
A majority of healthcare professionals surveyed in the IMscin001 study found that the SC formulation is easy to administer and could save time compared with IV
Basel, 14 November 2023 - Roche (SIX: RO, ROG; OTCQX: RHHBY) announced today that the European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended the approval of subcutaneous (SC, or under the skin) Tecentriq® (atezolizumab). Tecentriq SC can be injected in approximately seven minutes, with most injections taking between four and eight minutes compared with 30-60 minutes for intravenous (IV) infusion, which can free up time for patients, healthcare teams and caregivers.1 The CHMP recommended Tecentriq SC for all indications in which Tecentriq IV has been previously approved, including certain types of lung, liver, bladder and breast cancer.2 A final decision on its approval is expected from the European Commission in the near future.
“Tecentriq has helped to treat more than 430,000 people diagnosed with some of the most aggressive forms of cancer,” said Levi Garraway, M.D., Ph.D., Roche’s Chief Medical Officer and Head of Global Product Development. “Subcutaneous administration offers a faster and more convenient alternative to IV infusion. The CHMP’s recommendation brings us a step closer to offering the first subcutaneous PD-L1 cancer immunotherapy treatment to patients in the EU.”
The CHMP’s positive opinion is based on pivotal data from the Phase IB/III IMscin001 study, which showed comparable levels of Tecentriq in the blood, when administered subcutaneously, and a safety and efficacy profile consistent with the IV formulation.3 Roche recently presented mature overall survival (OS) data with a median follow-up of 9.5 months at the European Society for Medical Oncology (ESMO) Congress 2023. The updated analysis confirmed the earlier results and showed that OS and other efficacy endpoints were consistent between the SC and IV treatment arms.1 A majority of healthcare professionals who were surveyed as part of the study agreed that the SC formulation is easy to administer (90%) and that it could save time for healthcare teams compared with the IV formulation (75%).1
Tecentriq SC, which recently received its first marketing authorisation in Great Britain, was developed to provide patients with an alternative to the IV administration of Tecentriq and the potential for treatment outside of the hospital setting. It is Roche’s fourth subcutaneous cancer therapy.4-6 Multiple oncology studies suggest that the majority of cancer patients generally prefer SC over IV administration due to reduced discomfort, ease of administration and shorter duration of treatment.
IMscin001 is a Phase IB/III, global, multicentre, randomised study evaluating the pharmacokinetics, safety and efficacy of Tecentriq SC, compared with Tecentriq IV, in patients with previously treated locally advanced or metastatic non-small cell lung cancer (NSCLC) for whom prior platinum therapy has failed. The study enrolled 371 patients.
Part 2 of the study met its primary endpoints, demonstrating comparable levels of Tecentriq in the blood during a given dosing interval on the basis of established pharmacokinetic measurements; observed serum Ctrough and model-predicted area under the curve. Efficacy, as measured by progression-free survival (PFS), objective response rates (ORR) and OS, was similar between the SC and IV treatment arms and consistent with the known profile of Tecentriq IV. The safety profile of Tecentriq SC was also consistent with that of Tecentriq IV.
Tecentriq SC combines Tecentriq with Halozyme Therapeutics’ Enhanze® drug delivery technology.
Tecentriq is a monoclonal antibody designed to bind with a protein called programmed death ligand-1 (PD-L1), which is expressed on tumour cells and tumour-infiltrating immune cells, blocking its interactions with both PD-1 and B7.1 receptors. By inhibiting PD-L1, Tecentriq may enable the activation of T-cells. Tecentriq is a cancer immunotherapy that has the potential to be used as a foundational combination partner with other immunotherapies, targeted medicines and various chemotherapies across a broad range of cancers.
The Enhanze drug delivery technology is based on a proprietary recombinant human hyaluronidase PH20 (rHuPH20), an enzyme that locally and temporarily degrades hyaluronan – a glycosaminoglycan or chain of natural sugars in the body – in the subcutaneous space. This increases the permeability of the tissue under the skin, allowing space for Tecentriq to enter, enabling it to be rapidly dispersed and absorbed into the bloodstream.
Tecentriq is approved for some of the most aggressive and difficult-to-treat forms of cancer. Tecentriq was the first cancer immunotherapy approved for the treatment of a certain type of early-stage (adjuvant) NSCLC, small cell lung cancer (SCLC) and hepatocellular carcinoma (HCC). Tecentriq is also approved in countries around the world, either alone or in combination with targeted therapies and/or chemotherapies, for various forms of metastatic NSCLC, certain types of metastatic urothelial cancer (mUC), PD-L1-positive metastatic triple-negative breast cancer (TNBC), BRAF V600 mutation-positive advanced melanoma and alveolar soft part sarcoma (ASPS).
To learn more about Roche’s scientific-led approach to cancer immunotherapy, please follow this link: https://www.roche.com/solutions/focus-areas/oncology/cancer-immunotherapy
Roche is a global pioneer in pharmaceuticals and diagnostics focused on advancing science to improve people’s lives. The combined strengths of pharmaceuticals and diagnostics under one roof have made Roche the leader in personalised healthcare – a strategy that aims to fit the right treatment to each patient in the best way possible.
Roche is the world’s largest biotech company, with truly differentiated medicines in oncology, immunology, infectious diseases, ophthalmology and diseases of the central nervous system. Roche is also the world leader in in vitro diagnostics and tissue-based cancer diagnostics, and a frontrunner in diabetes management.
Founded in 1896, Roche continues to search for better ways to prevent, diagnose and treat diseases and make a sustainable contribution to society. The company also aims to improve patient access to medical innovations by working with all relevant stakeholders. More than thirty medicines developed by Roche are included in the World Health Organization Model Lists of Essential Medicines, among them life-saving antibiotics, antimalarials and cancer medicines. Moreover, for the twelfth consecutive year, Roche has been recognised as one of the most sustainable companies in the Pharmaceuticals Industry by the Dow Jones Sustainability Indices (DJSI).
The Roche Group, headquartered in Basel, Switzerland, is active in over 100 countries and in 2020 employed more than 100,000 people worldwide. In 2020, Roche invested CHF 12.2 billion in R&D and posted sales of CHF 58.3 billion. Genentech, in the United States, is a wholly owned member of the Roche Group. Roche is the majority shareholder in Chugai Pharmaceutical, Japan
Elevidys-treated boys aged 4-7 years with Duchenne showed an increase on the North Star Ambulatory Assessment (NSAA), a measure of motor function, compared to placebo at 52 weeks but the primary endpoint was not met
For all key pre-specified secondary functional endpoints, time to rise and 10-metre walk test across age groups, clinically meaningful and statistically significant treatment benefits were observed
No new safety signals observed, reinforcing the favourable and manageable safety profile observed with Elevidys to date
Further evaluation of data is ongoing and Roche will discuss the path forward with health authorities
Roche (SIX: RO, ROG; OTCQX: RHHBY) announced today topline results from the global, randomised, double-blind Phase 3 EMBARK study of Elevidys™ (delandistrogene moxeparvovec) in ambulatory boys (those who can walk) with Duchenne muscular dystrophy aged 4-7 years. In the study, Elevidys-treated patients improved 2.6 points on their NSAA total score 52 weeks after treatment, compared to 1.9 points in placebo-treated patients (0.65; n=125; P=0.24).
In all pre-specified, timed functional key secondary endpoints, time to rise from floor and 10 metre walk test, clinically meaningful and statistically significant improvements were observed. Both endpoints are prognostic factors for disease progression and loss of ability to walk. Additionally, a clinically meaningful and statistically significant improvement was also observed for the pre-specified secondary endpoint stride velocity 95th centile. This novel digital endpoint, qualified by the European Medicines Agency (EMA), measures speed of walking via a wearable device (Syde®). The time to ascend 4-steps secondary endpoint also demonstrated consistent treatment benefit in favour of Elevidys.
All data are being further analysed and will be discussed with health authorities to determine the path forward. Detailed results from the EMBARK study will be shared at an upcoming scientific congress and a medical journal publication will be pursued.
“High unmet need remains in Duchenne and we are encouraged by the consistent and meaningful results seen in all key secondary functional endpoints for Elevidys, an innovative gene therapy,” said Levi Garraway, M.D., Ph.D., Chief Medical Officer and Head of Global Product Development, Roche. “We will work to further analyse the EMBARK results and consult with health authorities as quickly as possible. We sincerely thank all the boys, their families and the wider Duchenne community involved in this important research effort.”
All key pre-specified functional secondary endpoints demonstrated robust evidence for a clinically meaningful treatment benefit that was consistent across age groups in Elevidys-treated patients compared to placebo at week 52. These include:
Functional endpoints: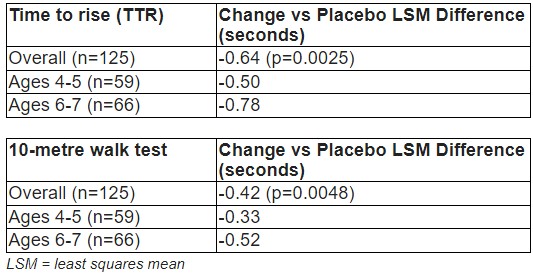
As part of a collaboration agreement Roche is working with Sarepta Therapeutics to transform the future for the Duchenne community, enabling those living with the disease to maintain and protect their muscle function, keeping them stronger for longer. Sarepta is responsible for managing regulatory approval and the commercialisation of Elevidys in the US. Roche is responsible for regulatory approvals and bringing Elevidys to patients across the rest of the world. Sarepta is responsible for the manufacturing of Elevidys and together, the companies are working on a comprehensive joint clinical development plan to maximise the chances of broad approval and access.
Study 101 evaluating the safety of Elevidys in four ambulatory participants aged between 4-<8 years old with Duchenne. Four-year data show a durable response and consistent safety profile.
Study 102, a Phase 2 clinical trial evaluating the safety and efficacy of Elevidys in patients with Duchenne aged 4-<8 years.
Study 103 (ENDEAVOR), a two-part, open-label, Phase 1b study assessing the Elevidys-dystrophin expression and safety of Elevidys in five cohorts of boys with Duchenne representing different stages of disease progression. This study is ongoing.
Study 301 (EMBARK), a Phase 3 global, randomised, double-blinded and placebo-controlled study of Elevidys in ambulatory Duchenne patients aged 4-<8 years old. The ENVOL trial (Study 302) a Phase 2 study in children with Duchenne. The study aims to enrol 21 participants who are under 4 years of age, including newborns. Not yet started.
The ENVISION trial (Study 303), a Phase 3 study in older ambulatory/non-ambulatory patients which is now recruiting.br/>
The EXPEDITION long-term (5 year) follow up study (Study 305) of participants who have received Elevidys in a previous clinical study, which is not yet recruiting.
EMBARK is a multinational, Phase 3, randomised, double-blind, two-part crossover, placebo-controlled study assessing the safety and efficacy of Elevidys in ambulatory boys with a confirmed mutation in the DMD gene, aged between 4 and 7 years. Eligible participants received a single dose of Elevidys during either Part 1 or Part 2 of the study. The study is ongoing.
Participants (n=125) received 1.33x1014 vg/kg of delandistrogene moxeparvovec or placebo. In Part 1, participants were randomised according to age (4-5 or 6-7) or NSAA total score at screening (≤22 or >22) to receive either Elevidys or placebo, with a follow-up period for 52 weeks. In Part 2, participants crossed over - meaning, those who were previously treated with placebo in Part 1 received Elevidys and participants who were previously treated with placebo received Elevidys, with a follow-up period for 52 weeks.
The primary endpoint of the trial was change from baseline in NSAA total score at week 52. Secondary endpoints include:
The quantity of delandistrogene moxeparvovec micro-dystrophin expression at Week 12 as measured by western blot of biopsied muscle tissue (Part 1)
Change from baseline to Week 52 in Time to Rise from Floor
Change from baseline to Week 52 in 10-metre Walk/Run (10MWR)
Change from baseline to Week 52 in stride velocity 95th centile (as measured by Syde®, a wearable device)
Change from baseline to Week 52 in 100-metre Walk/Run
Change from baseline to Week 52 in time to ascend 4 steps
Data not yet available for the following endpoints:
Change from baseline to Week 52 in Patient-Reported Outcomes Measurement Information System® (PROMIS®) mobility score
Change from baseline to Week 52 in PROMIS® upper extremity score
Number of skills gained or improved at Week 52 as measured by the NSAA
Elevidys™ (delandistrogene moxeparvovec, also known as SRP-9001) is the first approved disease-modifying therapy for Duchenne and is designed to address the underlying cause of Duchenne through targeted skeletal, respiratory and cardiac muscle expression of shortened dystrophin produced by Elevidys. Elevidys is a one-time treatment administered through a single (one-time) intravenous dose. Elevidys is contraindicated in patients with any deletion in exons 8 and/or 9 in the DMD gene.
Elevidys received accelerated approval in the US in June 2023, in the United Arab Emirates in August 2023 and in Qatar in September 2023 for the treatment of ambulant children aged 4 through 5 years with Duchenne, who have a confirmed mutation in the DMD gene.
Duchenne is a rare, genetic, muscle-wasting disease that progresses rapidly from early childhood. Approximately 1 in 3,500 - 5,000 boys worldwide are born with Duchenne, while Duchenne in girls is very rare. Everyone who has Duchenne will lose the ability to walk, upper limb, lung and cardiac function and mean life expectancy is 28 years. A diagnosis of DMD will require full-time caregiving which is most often provided by parents, the majority of whom will find it difficult to carry out usual work or household activities and suffer from depression and physical pain.
Duchenne is caused by mutations of the DMD gene, which affects the production of the muscle protein, dystrophin. Dystrophin is a critical component of a protein complex that strengthens muscle fibers and protects them from injury during muscle contraction. Due to a genetic mutation in the DMD gene, people with Duchenne do not make functional dystrophin; their muscle cells are more sensitive to injury and muscle tissue is progressively replaced with scar tissue and fat.
Neuroscience is a major focus of research and development at Roche. Our goal is to pursue groundbreaking science to develop new treatments that help improve the lives of people with chronic and potentially devastating diseases.
Roche is investigating more than a dozen medicines for neurological disorders, including neuromuscular diseases: Duchenne muscular dystrophy, facioscapulohumeral muscular dystrophy, myasthenia gravis and spinal muscular atrophy; neuro immune diseases: multiple sclerosis and neuromyelitis optica spectrum disorder; and neurodegenerative diseases: Alzheimer’s disease, Huntington’s disease and Parkinson’s disease. Together with our partners, we are committed to pushing the boundaries of scientific understanding to solve some of the most difficult challenges in neuroscience today.
Roche is a global pioneer in pharmaceuticals and diagnostics focused on advancing science to improve people’s lives. The combined strengths of pharmaceuticals and diagnostics under one roof have made Roche the leader in personalised healthcare – a strategy that aims to fit the right treatment to each patient in the best way possible.
Roche is the world’s largest biotech company, with truly differentiated medicines in oncology, immunology, infectious diseases, ophthalmology and diseases of the central nervous system. Roche is also the world leader in in vitro diagnostics and tissue-based cancer diagnostics, and a frontrunner in diabetes management.
Founded in 1896, Roche continues to search for better ways to prevent, diagnose and treat diseases and make a sustainable contribution to society. The company also aims to improve patient access to medical innovations by working with all relevant stakeholders. More than thirty medicines developed by Roche are included in the World Health Organization Model Lists of Essential Medicines, among them life-saving antibiotics, antimalarials and cancer medicines. Moreover, for the twelfth consecutive year, Roche has been recognised as one of the most sustainable companies in the Pharmaceuticals Industry by the Dow Jones Sustainability Indices (DJSI).
The Roche Group, headquartered in Basel, Switzerland, is active in over 100 countries and in 2020 employed more than 100,000 people worldwide. In 2020, Roche invested CHF 12.2 billion in R&D and posted sales of CHF 58.3 billion. Genentech, in the United States, is a wholly owned member of the Roche Group. Roche is the majority shareholder in Chugai Pharmaceutical, Japan
RVO is the third indication for Vabysmo, in addition to neovascular or ‘wet’ age-related macular degeneration and diabetic macular edema
Approval is based on two phase III studies demonstrating early and sustained vision improvements that were non-inferior to aflibercept
Vabysmo also demonstrated rapid and robust drying of retinal fluid
Additional U.S. label update across indications includes information on rare post-marketing reports of retinal vasculitis and/or retinal vascular occlusion; reporting rate is in line with other broadly used intravitreal treatments
Basel, 27 October 2023 - Roche (SIX: RO, ROG; OTCQX: RHHBY) announced today that the United States Food and Drug Administration (U.S. FDA) has approved Vabysmo® (faricimab) for the treatment of macular edema following retinal vein occlusion (RVO). RVO is the third indication for Vabysmo, in addition to neovascular or ‘wet’ age-related macular degeneration (nAMD) and diabetic macular edema (DME).1 Together, the three retinal conditions affect around 70 million people worldwide and are among the leading causes of vision loss.2-5
“Vabysmo is a new treatment option for RVO that can help people preserve and improve their vision, with the added benefit of retinal drying,” said Levi Garraway, M.D., Ph.D., Roche’s Chief Medical Officer and Head of Global Product Development. “The efficacy and safety profile of Vabysmo has been well established in global clinical trials and is reinforced by a growing breadth of real-world evidence, with hundreds of thousands of people treated.”
Vabysmo is the first and only bispecific antibody approved for the eye.1,6 Today’s approval in RVO is based on positive results from the global phase III BALATON and COMINO studies that demonstrated monthly treatment with Vabysmo provided early and sustained improvement in vision in people with branch and central RVO, meeting the primary endpoint of non-inferior visual acuity gains at 24 weeks compared to aflibercept. This was further supported by data showing Vabysmo achieved rapid and robust drying of retinal fluid. In BALATON and COMINO, Vabysmo was generally well tolerated and the safety profile was consistent with previous trials. The most common adverse reaction was conjunctival haemorrhage (3%). Safety results were consistent across study arms.7-9
Information has also been added to the Warnings and Precautions section of the U.S. label based on rare post-marketing cases of retinal vasculitis and/or retinal vascular occlusion, typically in the presence of intraocular inflammation. The reported rate of retinal vasculitis with vascular occlusion is 0.06 per 10,000 injections, in line with real-world reported frequencies of other broadly used intravitreal treatments for people living with nAMD, DME and RVO.10-12
To date, Vabysmo is approved in more than 80 countries around the world for people living with nAMD and DME, with approximately 2 million doses distributed globally
RVO is the second most common cause of vision loss due to retinal vascular conditions. It affects an estimated 28 million adults globally, mainly those aged 60 or older, and can lead to severe and sudden vision loss.2,13 The level of angiopoietin-2 (Ang-2) is elevated in RVO and it is thought that increased Ang-2 expression drives disease progression.14,15 RVO typically results in sudden, painless vision loss in the affected eye because the vein blockage restricts normal blood flow in the affected retina, resulting in ischaemia, bleeding, fluid leakage and retinal swelling called macular edema.13,16,17 Currently, macular edema due to RVO is typically treated with repeated intravitreal injections of anti-vascular endothelial growth factor therapies.16 There are two main types of RVO: branch retinal vein occlusion, which affects more than 23 million people globally and occurs when one of the four smaller ‘branches’ of the main central retinal vein becomes blocked; and central retinal vein occlusion, which is less common, affecting more than four million people worldwide, and occurs when the eye’s central retinal vein becomes blocked.
BALATON (NCT04740905) and COMINO (NCT04740931) are two randomised, multicentre, double-masked, global phase III studies evaluating the efficacy and safety of Vabysmo®️ (faricimab) compared to aflibercept. For the first 20 weeks, patients were randomised 1:1 to receive monthly injections for six months of either Vabysmo (6.0 mg) or aflibercept (2.0 mg). From weeks 24 to 72, all patients received Vabysmo (6.0 mg) up to every four months, using a treat-and-extend dosing regimen.
The BALATON study was conducted in 553 people with branch retinal vein occlusion. The COMINO study was conducted in 729 people with central retinal or hemiretinal vein occlusion. The primary endpoint of each study was the change in best-corrected visual acuity from baseline at 24 weeks. Secondary endpoints (weeks 0-24) included change in central subfield thickness and drying of retinal fluid, from baseline over time up to week 24. Secondary endpoints (weeks 24-72) were treatment durability at 68 weeks and continuation of weeks 0-24 endpoints.
Roche has a robust phase III clinical development programme for Vabysmo. The programme includes AVONELLE-X, an extension study of TENAYA and LUCERNE, evaluating the long-term safety and tolerability of Vabysmo in neovascular or ‘wet’ age-related macular degeneration (nAMD), and RHONE-X, an extension study of YOSEMITE and RHINE, evaluating the long-term safety and tolerability of Vabysmo in diabetic macular edema (DME).18,19 Roche has also initiated several phase IV studies, including the ELEVATUM study of Vabysmo in underrepresented patient populations with DME, the SALWEEN study of Vabysmo in a subpopulation of nAMD highly prevalent in Asia, as well as the VOYAGER study, a global real-world data collection platform.20-22 Roche also supports several other independent studies to further understand retinal conditions with a high unmet need.
Vabysmo is the first bispecific antibody approved for the eye.1,6 It targets and inhibits two signalling pathways linked to a number of vision-threatening retinal conditions by neutralising angiopoietin-2 (Ang-2) and vascular endothelial growth factor-A (VEGF-A). Ang-2 and VEGF-A contribute to vision loss by destabilising blood vessels, causing new leaky blood vessels to form and increasing inflammation. By blocking pathways involving Ang-2 and VEGF-A, Vabysmo is designed to stabilise blood vessels.23,24 Vabysmo is approved in more than 80 countries around the world, including the United States, Japan, the United Kingdom and the European Union for people living with neovascular or ‘wet’ age-related macular degeneration and diabetic macular edema, and in the United States for people living with macular edema following retinal vein occlusion. Review by other regulatory authorities is ongoing
Roche is focused on saving people’s eyesight from the leading causes of vision loss through pioneering therapies. Through our innovation in the scientific discovery of new potential drug targets, personalised healthcare, molecular engineering, biomarkers and continuous drug delivery, we strive to design the right therapies for the right patients.
We have the broadest retina pipeline in ophthalmology, which is led by science and informed by insights from people with eye diseases. Our pipeline includes gene therapies and treatments for geographic atrophy and other vision-threatening diseases, including rare and inherited conditions.
Applying our extensive experience, we have already brought breakthrough ophthalmic treatments to people living with vision loss. Susvimo™ (previously called Port Delivery System with ranibizumab) 100 mg/mL for intravitreal use via ocular implant is the first United States Food and Drug Administration-approved refillable eye implant for neovascular or ‘wet’ age-related macular degeneration that continuously delivers a customised formulation of ranibizumab over a period of months.27 Vabysmo® (faricimab) is the first bispecific antibody approved for the eye, which targets and inhibits two signalling pathways linked to a number of vision-threatening retinal conditions.1,6,23,24 Lucentis® (ranibizumab injection)^ is the first treatment approved to improve vision in people with certain retinal conditions.
Roche is a global pioneer in pharmaceuticals and diagnostics focused on advancing science to improve people’s lives. The combined strengths of pharmaceuticals and diagnostics under one roof have made Roche the leader in personalised healthcare – a strategy that aims to fit the right treatment to each patient in the best way possible.
Roche is the world’s largest biotech company, with truly differentiated medicines in oncology, immunology, infectious diseases, ophthalmology and diseases of the central nervous system. Roche is also the world leader in in vitro diagnostics and tissue-based cancer diagnostics, and a frontrunner in diabetes management.
Founded in 1896, Roche continues to search for better ways to prevent, diagnose and treat diseases and make a sustainable contribution to society. The company also aims to improve patient access to medical innovations by working with all relevant stakeholders. More than thirty medicines developed by Roche are included in the World Health Organization Model Lists of Essential Medicines, among them life-saving antibiotics, antimalarials and cancer medicines. Moreover, for the twelfth consecutive year, Roche has been recognised as one of the most sustainable companies in the Pharmaceuticals Industry by the Dow Jones Sustainability Indices (DJSI).
The Roche Group, headquartered in Basel, Switzerland, is active in over 100 countries and in 2020 employed more than 100,000 people worldwide. In 2020, Roche invested CHF 12.2 billion in R&D and posted sales of CHF 58.3 billion. Genentech, in the United States, is a wholly owned member of the Roche Group. Roche is the majority shareholder in Chugai Pharmaceutical, Japan
Vabysmo achieved its primary endpoint of non-inferiority to aflibercept in RVO in the BALATON and COMINO clinical trials
Vabysmo was generally well tolerated, with a safety profile consistent with previous trials
Vabysmo is the first and only treatment that targets and inhibits two disease pathways involving Ang-2 and VEGF-A, linked to a number of vision-threatening retinal conditions
Detailed results will be presented at an upcoming medical meeting and submitted to regulatory authorities around the world
Basel, 27 October 2022 - Roche (SIX: RO, ROG; OTCQX: RHHBY) today announced positive topline results from two global phase III studies, BALATON and COMINO, evaluating the first and only bispecific antibody for the eye, Vabysmo® (faricimab), in macular edema due to branch and central retinal vein occlusion (BRVO and CRVO).1,2,3 RVO is a vision-threatening condition that impacts 28 million people globally.
Both studies met their primary endpoints, showing that people with macular edema due to BRVO and CRVO receiving Vabysmo injections every four weeks, for up to 24 weeks, achieved non-inferior visual acuity gains compared to those receiving aflibercept injections every four weeks.
“These encouraging data demonstrate that Vabysmo could potentially provide a new treatment option for people living with retinal vein occlusion, a serious retinal vascular condition that can lead to irreversible vision impairment or vision loss,” said Levi Garraway, M.D., Ph.D., Roche’s Chief Medical Officer and Head of Global Product Development. “Today’s results add to the extensive evidence supporting Vabysmo’s efficacy in treating multiple types of retinal conditions. We look forward to submitting these data to regulatory authorities.”
Vabysmo also showed rapid drying of retinal fluid from baseline through week 24, as measured by reduction in central subfield thickness.
In both studies, Vabysmo was generally well tolerated. The safety profile was consistent with previous trials.
Detailed results will be presented at an upcoming medical meeting and submitted to regulatory authorities around the world.
Vabysmo is uniquely engineered to target and inhibit two disease pathways, which are linked to a number of vision-threatening retinal conditions, by neutralising angiopoietin-2 (Ang-2) and vascular endothelial growth factor-A (VEGF-A) to restore vascular stability.3,5 The level of Ang-2 is elevated in RVO and it is thought that increased Ang-2 expression drives disease progression.
To date, Vabysmo is approved in more than 40 countries around the world, including the United States, Japan, the United Kingdom and the European Union, for people living with neovascular or ‘wet’ age-related macular degeneration (nAMD) and diabetic macular edema (DME).8,9,10,11,12 Vabysmo’s long-term efficacy and safety in nAMD and DME has been demonstrated by two-year data from four large, global studies involving more than 3,000 participants.3,5,13,14 Vabysmo is the only injectable eye medicine approved with phase III studies supporting treatment intervals of up to four months for people living with nAMD and DME.12 Globally, more than 165,000 Vabysmo doses have been distributed for treatment of these conditions to date.8 RVO, nAMD and DME together affect around 70 million people worldwide and are among the leading causes of vision loss.
RVO is the second most common cause of vision loss due to retinal vascular diseases.4 It affects an estimated 28 million adults globally, mainly those aged 60 or older, and can lead to severe and sudden vision loss.4,18 The level of angiopoietin-2 (Ang-2) is elevated in RVO and it is thought that increased Ang-2 expression drives disease progression.6,7 RVO typically results in sudden, painless vision loss in the affected eye because the vein blockage restricts normal blood flow in the affected retina, resulting in ischemia, bleeding, fluid leakage and retinal swelling called macular edema.18,19,20 Currently, macular edema due to RVO is typically treated with repeated intravitreal injections of anti-vascular endothelial growth factor therapies.20 There are two main types of RVO: branch retinal vein occlusion, which affects more than 23 million people globally and occurs when one of the four smaller ‘branches’ of the main central retinal vein becomes blocked; and central retinal vein occlusion, which is less common, affecting more than four million people worldwide, and occurs when the eye’s central retinal vein becomes blocked.
BALATON (NCT04740905) and COMINO (NCT04740931) are two randomised, multicentre, double-masked, global phase III studies evaluating the efficacy and safety of Vabysmo®️ (faricimab) compared to aflibercept. For the first 20 weeks, patients are randomised 1:1 to receive six monthly injections of either Vabysmo (6.0 mg) or aflibercept (2.0 mg). From weeks 24-72, all patients receive Vabysmo (6.0 mg) up to every four months – according to a personalised treatment interval dosing regimen – using a treat-and-extend approach.
The BALATON study is being conducted in 553 people with branch retinal vein occlusion. The COMINO study is being conducted in 729 people with central retinal or hemiretinal vein occlusion.
The primary endpoint of each study is the change in best-corrected visual acuity from baseline at 24 weeks. Secondary endpoints include change in central subfield thickness and drying of retinal fluid from baseline over time up to week 24.
Roche has a robust phase III clinical development programme for Vabysmo. The programme includes AVONELLE-X, an extension study of TENAYA and LUCERNE, evaluating the long-term safety and tolerability of Vabysmo in neovascular or ‘wet’ age-related macular degeneration, and RHONE-X, an extension study of YOSEMITE and RHINE evaluating the long-term safety and tolerability of Vabysmo in diabetic macular edema (DME).21,22 Roche has also initiated the phase IV ELEVATUM study of Vabysmo in underrepresented patient populations with DME and supports several other independent studies to further understand retinal conditions with a high unmet need.
Vabysmo is the first bispecific antibody approved for the eye.9,11 It targets and inhibits two disease pathways linked to a number of vision-threatening retinal conditions by neutralising angiopoietin-2 (Ang-2) and vascular endothelial growth factor-A (VEGF-A). Ang-2 and VEGF-A contribute to vision loss by destabilising blood vessels, causing new leaky blood vessels to form and increasing inflammation.3,5 By blocking pathways involving Ang-2 and VEGF-A, Vabysmo is designed to stabilise blood vessels.3,5 Vabysmo is approved in more than 40 countries around the world, including the United States, Japan, the United Kingdom and the European Union for people living with neovascular or ‘wet’ age-related macular degeneration and diabetic macular edema. Review by other regulatory authorities is ongoing.
Roche is focused on saving people’s eyesight from the leading causes of vision loss through pioneering therapies. Through our innovation in the scientific discovery of new potential drug targets, personalised healthcare, molecular engineering, biomarkers and continuous drug delivery, we strive to design the right therapies for the right patients.
We have the broadest retina pipeline in ophthalmology, which is led by science and informed by insights from people with eye diseases. Our pipeline includes gene therapies and treatments for geographic atrophy and other vision-threatening diseases, including rare and inherited conditions.
Applying our extensive experience, we have already brought breakthrough ophthalmic treatments to people living with vision loss. Susvimo™ (previously called Port Delivery System with ranibizumab) 100 mg/mL for intravitreal use via ocular implant is the first United States Food and Drug Administration-approved refillable eye implant for neovascular or ‘wet’ age-related macular degeneration that continuously delivers a customised formulation of ranibizumab over a period of months.24 Vabysmo® (faricimab) is the first bispecific antibody approved for the eye, which targets two disease pathways that drive retinal conditions.3,5,9,11 Lucentis® (ranibizumab injection) is the first treatment approved to improve vision in people with certain retinal conditions.
Roche is a global pioneer in pharmaceuticals and diagnostics focused on advancing science to improve people’s lives. The combined strengths of pharmaceuticals and diagnostics under one roof have made Roche the leader in personalised healthcare – a strategy that aims to fit the right treatment to each patient in the best way possible.
Roche is the world’s largest biotech company, with truly differentiated medicines in oncology, immunology, infectious diseases, ophthalmology and diseases of the central nervous system. Roche is also the world leader in in vitro diagnostics and tissue-based cancer diagnostics, and a frontrunner in diabetes management.
Founded in 1896, Roche continues to search for better ways to prevent, diagnose and treat diseases and make a sustainable contribution to society. The company also aims to improve patient access to medical innovations by working with all relevant stakeholders. More than thirty medicines developed by Roche are included in the World Health Organization Model Lists of Essential Medicines, among them life-saving antibiotics, antimalarials and cancer medicines. Moreover, for the twelfth consecutive year, Roche has been recognised as one of the most sustainable companies in the Pharmaceuticals Industry by the Dow Jones Sustainability Indices (DJSI).
The Roche Group, headquartered in Basel, Switzerland, is active in over 100 countries and in 2020 employed more than 100,000 people worldwide. In 2020, Roche invested CHF 12.2 billion in R&D and posted sales of CHF 58.3 billion. Genentech, in the United States, is a wholly owned member of the Roche Group. Roche is the majority shareholder in Chugai Pharmaceutical, Japan
77% of early-stage relapsing-remitting multiple sclerosis (RRMS) patients who had not received prior treatment achieved no evidence of disease activity (NEDA) at two years
Initiation of OCREVUS as first-line treatment reduces relapses, hospitalisations and costs compared with using OCREVUS in second-line setting
Nine-year long-term safety data for OCREVUS further reinforce favourable benefit-risk profile; more than 250,000 people have been treated globally
Pregnancy outcomes reported for more than 2,000 women with multiple sclerosis (MS) treated with OCREVUS do not suggest an increased risk of adverse pregnancy and infant outcomes
Basel, 26 October 2022 - Roche (SIX: RO, ROG; OTCQX: RHHBY) today announced new OCREVUS® (ocrelizumab) data on disease progression and healthcare costs in patients with early-stage RRMS and long-term safety from all clinical trials in patients with relapsing MS (RMS) and primary progressive MS (PPMS). Data from the largest database of pregnancy outcomes for an anti-CD20 therapy in MS suggest consistent outcomes with epidemiological data in pregnant women and was an oral presentation today at the 38th Congress of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS).
“MS often impacts young people at a time in their lives when they are starting a career or planning a family,” said Levi Garraway, M.D., Ph.D. Roche's Chief Medical Officer and Head of Global Product Development. “These new data show that using OCREVUS as a first-line treatment brings substantial clinical and cost benefits to patients, thereby further emphasizing the efficacy that OCREVUS may bring with continued long-term use.”
Two-year interim analysis of open-label Phase IIIb ENSEMBLE: No evidence of disease progression in early-stage RRMS
OCREVUS treatment provided consistent benefit over two years in patients who were recently diagnosed with RRMS and had not received prior disease modifying treatment (DMT) in an interim analysis of open-label Phase IIIb study ENSEMBLE. After 96 weeks of OCREVUS treatment, 77% of patients achieved no evidence of disease activity (NEDA; no relapses, no worsening of disability or no evidence of MRI lesion activity with pre-specified MRI re-baselining at 8 weeks). The majority of patients had no relapses (93%), no MRI lesion activity (89%) and no 24-week confirmed disability progression (91%).
Over two years, the average annualised relapse rate (ARR) across all patients in the ENSEMBLE study was low (0.033), which equates to 1 relapse every 30 years. The mean Expanded Disability Status Scale (EDSS) score from baseline significantly improved from 1.8 to 1.67 (p<0.0001). The safety profile of OCREVUS in this trial was consistent with its overall favourable safety profile.
U.S. claims analysis: Early initiation of OCREVUS may benefit both patients and the healthcare system
Patients who were newly diagnosed and initiated OCREVUS treatment had a lower rate of annualised events often associated with a relapse (EOAR; 0.36) compared with patients who initiated OCREVUS as a second-line or later treatment (0.51). Additionally, patients treated with first-line OCREVUS had lower hospitalisation rates within one year compared with patients treated with second-line or later OCREVUS (0.02 vs. 0.042, respectively).
Costs were also lower after first-line OCREVUS treatment, including the total annual non-DMT costs ($18,389 vs. $26,225, respectively) and MS-related/non-DMT costs ($8,837 vs. $14,758, respectively) compared with patients treated with second-line or later OCREVUS.
Non-DMT costs were defined as total costs for emergency department visits, inpatient care, outpatient care and non-DMT prescriptions. MS-related costs were defined similarly but could also be attributed to the disease.
The findings from the study suggest that the early initiation of OCR, instead of escalation from lower-efficacy DMTs, can provide benefits for both patients and the healthcare system.
These clinical and economic analyses were performed on U.S. commercial claims data between 1 January 2015 and 30 June 2021.
Long-term safety from OCREVUS clinical trials consistent for nine years
New safety data as of November 2021 will be presented, representing 5,848 patients with relapsing MS (RMS) and primary progressive MS (PPMS) and 25,153 patient-years of exposure to OCREVUS, across all OCREVUS clinical trials. These findings further demonstrate the consistently favourable benefit-risk profile of OCREVUS over nine years.
“Nine-year data presented at ECTRIMS in relapsing and primary progressive MS continue to show significant efficacy against disease activity and progression with a consistent long-term safety profile, which is very encouraging for patients living with this disease and their physicians,” said Stephen Hauser, M.D., chair of the Scientific Steering Committee of the OPERA studies and director of the Weill Institute for Neurosciences at the University of California, San Francisco. “OCREVUS has significantly changed the treatment paradigm for more than 250,000 people with MS since its approval more than five years ago.”
More than 250,000 people with MS have now been treated with OCREVUS globally, and data continue to show a consistent and favourable benefit-risk profile in clinical trial and real-world settings. OCREVUS is approved in 101 countries across North America, South America, the Middle East, Eastern Europe, as well as in Australia, Switzerland, the United Kingdom and the EU.
Roche safety data do not suggest increased risk of adverse pregnancy and infant outcomes in women treated with OCREVUS
As of 31 March 2022, 2,020 cumulative MS pregnancies were reported, of which 705 (35%) had in utero exposure to OCREVUS.
Of the 532 pregnancies with in utero exposure of OCREVUS that were also prospectively reported, 286 had known outcomes: 79% live births; 1% ectopic pregnancies; 12% therapeutic/elective abortions; 8% spontaneous abortions; 0.3% still birth.
In women living with MS and treated with OCREVUS who reported pregnancies, cumulative data do not suggest an increased risk of preterm birth, major congenital anomalies or other adverse outcomes and are consistent with epidemiological data, and in-line with previous reports, providing important information for women living with MS who are or may become pregnant.
Regulatory agencies advise the use of contraception while on treatment with OCREVUS, and for 6-12 months after the last dose. The benefit-risk of OCREVUS in mothers and infants is being prospectively assessed in two Phase IV studies, MINORE in pregnant women and SOPRANINO in lactating women, both of which are currently enrolling.
Multiple sclerosis (MS) is a chronic disease that affects more than 2.8 million people worldwide. MS occurs when the immune system abnormally attacks the insulation and support around nerve cells (myelin sheath) in the central nervous system (brain, spinal cord and optic nerves), causing inflammation and consequent damage. This damage can cause a wide range of symptoms, including muscle weakness, fatigue and difficulty seeing, and may eventually lead to disability. Most people with MS experience their first symptom between 20 and 40 years of age, making the disease the leading cause of non-traumatic disability in younger adults.
People with all forms of MS experience disease progression – permanent loss of nerve cells in the central nervous system and gradual worsening of disability – at the beginning of their disease even if their clinical symptoms aren’t apparent or don’t appear to be getting worse. Delays in diagnosis and treatment can negatively impact people with MS, in terms of their physical and mental health, and contribute to the negative financial impact on the individual and society. An important goal of treating MS is to slow, stop and ideally prevent the progression of disability as early as possible.
Relapsing-remitting MS (RRMS) is the most common form of the disease and is characterised by episodes of new or worsening signs or symptoms (relapses) followed by periods of recovery. Approximately 85% of people with MS are initially diagnosed with RRMS. The majority of people who are diagnosed with RRMS will eventually transition to secondary progressive MS (SPMS), in which they experience steadily worsening disability over time. Relapsing forms of MS (RMS) include people with RRMS and people with SPMS who continue to experience relapses. Primary progressive MS (PPMS) is a debilitating form of the disease marked by steadily worsening symptoms but typically without distinct relapses or periods of remission. Approximately 15% of people with MS are diagnosed with the primary progressive form of the disease. Until the FDA approval of OCREVUS, there had been no FDA-approved treatments for PPMS.
OCREVUS is the first and only therapy approved for both RMS (including RRMS and active, or relapsing SPMS, in addition to clinically isolated syndrome [CIS] in the U.S.) and PPMS. OCREVUS is a humanised monoclonal antibody designed to target CD20-positive B cells, a specific type of immune cell thought to be a key contributor to myelin (nerve cell insulation and support) and axonal (nerve cell) damage. This nerve cell damage can lead to disability in people with MS. Based on preclinical studies, OCREVUS binds to CD20 cell surface proteins expressed on certain B cells, but not on stem cells or plasma cells, suggesting that important functions of the immune system may be preserved. OCREVUS is administered by intravenous infusion every six months. The initial dose is given as two 300 mg infusions given two weeks apart. Subsequent doses are given as single 600 mg infusions.
OCREVUS is the first and only therapy approved for both RMS (including RRMS and active, or relapsing, secondary progressive MS [SPMS], in addition to clinically isolated syndrome [CIS] in the U.S.) and PPMS. OCREVUS is a humanised monoclonal antibody designed to target CD20-positive B cells, a specific type of immune cell thought to be a key contributor to myelin (nerve cell insulation and support) and axonal (nerve cell) damage. This nerve cell damage can lead to disability in people with MS. Based on preclinical studies, OCREVUS binds to CD20 cell surface proteins expressed on certain B cells, but not on stem cells or plasma cells, suggesting that important functions of the immune system may be preserved. OCREVUS is administered by intravenous infusion every six months. The initial dose is given as two 300 mg infusions given two weeks apart. Subsequent doses are given as single 600 mg infusions.
Neuroscience is a major focus of research and development at Roche. Our goal is to pursue ground-breaking science to develop new treatments that help improve the lives of people with chronic and potentially devastating diseases.
Roche has both approved and investigational medicines across multiple sclerosis, spinal muscular atrophy, neuromyelitis optica spectrum disorder, myasthenia gravis, Alzheimer’s disease, Huntington’s disease, Parkinson’s disease and Duchenne muscular dystrophy. Together with our partners, we are committed to pushing the boundaries of scientific understanding to solve some of the most difficult challenges in neuroscience today.
Roche is a global pioneer in pharmaceuticals and diagnostics focused on advancing science to improve people’s lives. The combined strengths of pharmaceuticals and diagnostics under one roof have made Roche the leader in personalised healthcare – a strategy that aims to fit the right treatment to each patient in the best way possible.
Roche is the world’s largest biotech company, with truly differentiated medicines in oncology, immunology, infectious diseases, ophthalmology and diseases of the central nervous system. Roche is also the world leader in in vitro diagnostics and tissue-based cancer diagnostics, and a frontrunner in diabetes management.
Founded in 1896, Roche continues to search for better ways to prevent, diagnose and treat diseases and make a sustainable contribution to society. The company also aims to improve patient access to medical innovations by working with all relevant stakeholders. More than thirty medicines developed by Roche are included in the World Health Organization Model Lists of Essential Medicines, among them life-saving antibiotics, antimalarials and cancer medicines. Moreover, for the twelfth consecutive year, Roche has been recognised as one of the most sustainable companies in the Pharmaceuticals Industry by the Dow Jones Sustainability Indices (DJSI).
The Roche Group, headquartered in Basel, Switzerland, is active in over 100 countries and in 2020 employed more than 100,000 people worldwide. In 2020, Roche invested CHF 12.2 billion in R&D and posted sales of CHF 58.3 billion. Genentech, in the United States, is a wholly owned member of the Roche Group. Roche is the majority shareholder in Chugai Pharmaceutical, Japan
These Phase III data are the first and only to show an improvement in disease-free survival in early-stage resected ALK-positive non-small cell lung cancer (NSCLC)
With about one in two people with early-stage NSCLC experiencing disease recurrence following surgery, despite adjuvant chemotherapy,1 more effective treatment options are urgently needed to provide the best chance for cure
Data are being presented as a late-breaking oral during the ESMO 2023 Presidential Symposium
Basel, 18 October 2023 - Roche (SIX: RO, ROG; OTCQX: RHHBY) announced today results from the primary analysis of the Phase III ALINA study demonstrating a statistically significant and clinically meaningful improvement in disease-free survival (DFS; primary endpoint). The study results showed that Alecensa® (alectinib) reduces the risk of disease recurrence or death by 76% (hazard ratio [HR]=0.24, 95% CI: 0.13-0.43, p<0.0001) compared with platinum-based chemotherapy in people with completely resected stage IB (tumour ≥4cm) to IIIA (UICC/AJCC 7th edition) anaplastic lymphoma kinase (ALK)-positive non-small cell lung cancer (NSCLC).3 A clinically meaningful improvement of central nervous system (CNS)-DFS was also observed (HR=0.22; 95% CI: 0.08-0.58).3 The safety and tolerability of Alecensa in this trial were consistent with previous trials in the metastatic setting and no unexpected safety findings were observed.3 Overall survival data were immature at the time of this analysis and follow-up is ongoing to report a more mature estimate.
The full results of ALINA are being presented as a late-breaking oral at the European Society of Medical Oncology (ESMO) Congress 2023 Presidential Symposium on Saturday 21 October 2023. These data will be submitted to global health authorities, including the U.S. Food and Drug Administration and the European Medicines Agency.
“By reducing the risk of recurrence or death of ALK-positive early-stage NSCLC by an unprecedented 76%, Alecensa can potentially alter the course of this disease as we aim to provide the best chance for cure,” said Levi Garraway, M.D., Ph.D., Roche’s Chief Medical Officer and Head of Global Product Development. “We urgently need to do more to help people with lung cancer, as about half of patients with early-stage NSCLC experience disease recurrence. We’re working with health authorities to bring Alecensa to patients in this setting as soon as possible.”
“These potentially practice-changing data reinforce the potential of Alecensa as a new standard of care in the ALK-positive early lung cancer setting where treatment options are currently extremely limited,” said Professor Benjamin Solomon, Medical Oncologist, Peter MacCallum Cancer Centre, Australia. “The magnitude of disease-free survival observed in this study could represent a paradigm shift in the way we manage early-stage ALK-positive lung cancer.”
Delaying disease progression is of particular importance for people with ALK-positive NSCLC, who are generally younger – usually around 55 – and are at higher risk of developing brain metastases than those with other types of NSCLC.4 Once the disease returns it often spreads to other parts of the body, at which point it is usually considered incurable.2,5 Comprehensive biomarker testing is essential to helping physicians secure a complete, personalised diagnosis and identify the right treatment for each patient.
Results from the primary analysis of the ALINA study showed median DFS was not yet reached for Alecensa compared with 41.3 months for chemotherapy (95% CI: 28.5, not evaluable [NE]) in patients with stage IB (tumour ≥4cm) to IIIA disease.3 Grade 3 or 4 adverse events (AEs) occurred in 30% of people receiving Alecensa, compared with 31% of those receiving chemotherapy.3 No Grade 5 events were observed in either treatment arm.3 For those receiving Alecensa, 5.5% of patients discontinued treatment due to AEs versus 12.5% in the chemotherapy arm.
The ALINA study [NCT03456076] is a Phase III, randomised, active-controlled, multicentre, open-label study evaluating the efficacy and safety of adjuvant Alecensa® (alectinib) compared with platinum-based chemotherapy in people with completely resected stage IB (tumour ≥4cm) to IIIA (UICC/AJCC 7th edition) anaplastic lymphoma kinase (ALK)-positive NSCLC. The study includes 257 patients who were randomly assigned to either the investigational or control treatment arm. The primary endpoint is disease-free survival. Secondary outcome measures include overall survival and percentage of patients with adverse events.
Alecensa is a highly selective, central nervous system-active, oral medicine created at Chugai, a member of the Roche Group, Kamakura Research Laboratories for people with non-small cell lung cancer (NSCLC) whose tumours are identified as anaplastic lymphoma kinase (ALK) positive. Alecensa is already approved in over 100 countries as an initial (first-line) and second-line treatment for ALK-positive, metastatic NSCLC, including in the United States, Europe, Japan and China.
Lung cancer is one of the leading causes of cancer death globally.6 Each year 1.8 million people die as a result of the disease; this translates into more than 4,900 deaths worldwide every day.6 Lung cancer can be broadly divided into two major types: non-small cell lung cancer (NSCLC) and small-cell lung cancer (SCLC). NSCLC is the most prevalent type, accounting for around 85% of all cases.7 Today, about half of all people with early lung cancer (45-76%, depending on disease stage) still experience a cancer recurrence following surgery, despite adjuvant chemotherapy.1 Treating lung cancer early, before it has spread, may help prevent the disease from returning and provide people with the best opportunity for a cure.
Lung cancer is a major area of focus and investment for Roche, and we are committed to developing new approaches, medicines and tests that can help people with this deadly disease. Our goal is to provide an effective treatment option for every person diagnosed with lung cancer. We currently have six approved medicines to treat certain kinds of lung cancer and more than ten medicines being developed to target the most common genetic drivers of lung cancer or to boost the immune system to combat the disease. Roche is committed to improving treatment of early-stage lung cancers to help increase the chance of cure for more people.
Roche is a global pioneer in pharmaceuticals and diagnostics focused on advancing science to improve people’s lives. The combined strengths of pharmaceuticals and diagnostics under one roof have made Roche the leader in personalised healthcare – a strategy that aims to fit the right treatment to each patient in the best way possible.
Roche is the world’s largest biotech company, with truly differentiated medicines in oncology, immunology, infectious diseases, ophthalmology and diseases of the central nervous system. Roche is also the world leader in in vitro diagnostics and tissue-based cancer diagnostics, and a frontrunner in diabetes management.
Founded in 1896, Roche continues to search for better ways to prevent, diagnose and treat diseases and make a sustainable contribution to society. The company also aims to improve patient access to medical innovations by working with all relevant stakeholders. More than thirty medicines developed by Roche are included in the World Health Organization Model Lists of Essential Medicines, among them life-saving antibiotics, antimalarials and cancer medicines. Moreover, for the twelfth consecutive year, Roche has been recognised as one of the most sustainable companies in the Pharmaceuticals Industry by the Dow Jones Sustainability Indices (DJSI).
The Roche Group, headquartered in Basel, Switzerland, is active in over 100 countries and in 2020 employed more than 100,000 people worldwide. In 2020, Roche invested CHF 12.2 billion in R&D and posted sales of CHF 58.3 billion. Genentech, in the United States, is a wholly owned member of the Roche Group. Roche is the majority shareholder in Chugai Pharmaceutical, Japan
Neonatal sepsis is a leading cause of death for newborns
Testing IL-6 can indicate a neonatal sepsis infection earlier than other biomarkers
Earlier diagnosis of neonatal sepsis can lead to improved outcomes and a reduction of long-term complications from sepsis
Basel, 18 October 2023 - Roche (SIX: RO, ROG; OTCQX: RHHBY) announced today that the Elecsys IL-6 immunoassay has become the first IL-6 test to have a certified claim for use in diagnosis of neonatal sepsis, in countries accepting the CE Mark.*
Elecsys IL-6 aids physicians in combating the impact of neonatal sepsis by facilitating an early diagnosis. Early and improved diagnosis can contribute to appropriate use of antibiotics, as well as potentially decreasing mortality rates and mitigating long-term consequences of sepsis.
“Sepsis is one of the leading causes of death in newborns and we need to do everything we can to prevent these deaths," said Matt Sause, CEO of Roche Diagnostics. "Receiving the first approval for IL-6 use with newborns, is a significant step forward in helping clinicians confidently diagnose neonatal sepsis earlier and save more lives.”
Neonatal sepsis is an infection involving the bloodstream within the first four weeks of life and results in high rates of morbidity and mortality. Neonatal sepsis can initially present with subtle signs, but can rapidly progress to multisystem organ failure. Early detection and prompt intervention are essential to prevent severe and life-threatening complications.
Testing IL-6 is suitable for early diagnosis of neonatal sepsis as it increases rapidly in response to infection, much earlier than other markers, making it a better early warning marker of inflammation, infection, or sepsis. With earlier diagnosis, clinicians can make earlier and more appropriate interventions to give neonates the best chance of a positive outcome. The Elecsys IL-6 can help to achieve this with the test only taking 18 minutes to run and only using a small amount of blood.
Sepsis is a condition that can be caused by bacteria, fungi or viruses in the blood and is the result of the body’s response to infection. Sepsis is defined as life-threatening organ dysfunction caused by a dysregulated host response to infection. 20% of deaths worldwide are sepsis-related and patient survival decreases by ~8% with each hour of delay before effective treatment.
The first 28 days of life (the neonatal period) are the most vulnerable time for child survival. Every year, an estimated 2.5 million neonates die in their first month of life, accounting for nearly one-half of deaths in children under 5 years of age. An estimated 375 000 neonatal deaths due to sepsis occurred globally in 2018, which represented 15% of all neonatal deaths, making sepsis one of the leading causes of newborn death.
Newborns often present with non-specific signs and symptoms, which means an early diagnosis of neonatal sepsis can be difficult. Some laboratory tests used in sepsis diagnosis have a low sensitivity, particularly in the early phase of infection. It is also difficult to collect sufficient blood volumes from neonates, especially low birthweight infants, so using a small sample size is important to be minimally invasive to the baby. If a sufficient blood sample cannot be taken, it may lead to low positivity rate in blood cultures. In addition, blood culture results can take up to 48 hours, therefore treatment is often started before results are known.
The prognosis of neonatal sepsis depends on early recognition and appropriate treatment, although signs and symptoms are often nonspecific and may overlap with those of other severe conditions, such as meningitis and pneumonia. These clinical signs include respiratory distress and cyanosis, apnoea, feeding difficulties, lethargy or irritability, and poor perfusion.
Elecsys IL-6 immunoassay is an in vitro diagnostic test for the quantitative determination of IL-6 (interleukin-6) in human serum and plasma. This assay is used to assist in identifying severe inflammatory responses in patients. A test takes 18 minutes to run and only needs a sample volume of 30 μL (cobas® e411, e601, e602); or 18 μL (cobas® e402, e801), one of the smallest volumes on the market. The Elecsys IL-6 immunoassay is an electrochemiluminescence immunoassay “ECLIA” and is intended for use on cobas e immunoassay analyzers.
Roche is a global pioneer in pharmaceuticals and diagnostics focused on advancing science to improve people’s lives. The combined strengths of pharmaceuticals and diagnostics under one roof have made Roche the leader in personalised healthcare – a strategy that aims to fit the right treatment to each patient in the best way possible.
Roche is the world’s largest biotech company, with truly differentiated medicines in oncology, immunology, infectious diseases, ophthalmology and diseases of the central nervous system. Roche is also the world leader in in vitro diagnostics and tissue-based cancer diagnostics, and a frontrunner in diabetes management.
Founded in 1896, Roche continues to search for better ways to prevent, diagnose and treat diseases and make a sustainable contribution to society. The company also aims to improve patient access to medical innovations by working with all relevant stakeholders. More than thirty medicines developed by Roche are included in the World Health Organization Model Lists of Essential Medicines, among them life-saving antibiotics, antimalarials and cancer medicines. Moreover, for the twelfth consecutive year, Roche has been recognised as one of the most sustainable companies in the Pharmaceuticals Industry by the Dow Jones Sustainability Indices (DJSI).
The Roche Group, headquartered in Basel, Switzerland, is active in over 100 countries and in 2020 employed more than 100,000 people worldwide. In 2020, Roche invested CHF 12.2 billion in R&D and posted sales of CHF 58.3 billion. Genentech, in the United States, is a wholly owned member of the Roche Group. Roche is the majority shareholder in Chugai Pharmaceutical, Japan
Late-breaking Phase III results show subcutaneous injection was non-inferior to intravenous infusion based on OCREVUS levels in the blood over 12 weeks
OCREVUS subcutaneous injection was comparable to IV infusion in providing rapid and sustained depletion of B cells and near-complete suppression of MRI lesion activity in the brain over 24 weeks
The safety profile of OCREVUS subcutaneous injection was consistent with the well-established safety profile of OCREVUS IV infusion
The 10-minute subcutaneous injection has potential to improve the treatment experience and expand usage for people with multiple sclerosis (MS) in centres with IV capacity limitations
Roche (SIX: RO, ROG; OTCQX: RHHBY) today announced late-breaking data from the Phase III OCARINA II study. Study results demonstrate the effect of OCREVUS® (ocrelizumab) as an investigational twice-yearly, 10-minute subcutaneous injection on pharmacokinetic, biomarker, and MRI measures in patients with relapsing or primary progressive multiple sclerosis (RMS or PPMS). The data will be presented in a poster at the 9th Joint ECTRIMS-ACTRIMS Meeting (European and Americas Committees for Treatment and Research in Multiple Sclerosis).
“We are pleased to share that OCREVUS 10-minute subcutaneous injection suppressed brain lesions as effectively as the intravenous infusion,” said Levi Garraway, M.D., Ph.D., Roche’s Chief Medical Officer and Head of Global Product Development. “Having this additional treatment option may improve the treatment experience for both patients and physicians, and we hope the twice-a-year dosing will offer the same high adherence and persistence.’’
OCREVUS subcutaneous injection was non-inferior to OCREVUS IV infusion as measured by OCREVUS levels in the blood of patients (area under the serum concentration time curve) from day 1 to 12 weeks (3500 day*µg/mL for subcutaneous injection vs. 2750 day*µg/mL for IV infusion). Peak OCREVUS blood (serum) concentrations were similar for subcutaneous injection (132 µg/mL) and IV infusion (137 µg/mL).
OCREVUS subcutaneous injection provided rapid, sustained and near-complete B-cell depletion that was similar to OCREVUS IV infusion (97% and 98% of patients respectively had B cells levels of 5 cells/µL or less when first measured at 14 days), which was sustained over 24 weeks. At the time of analysis, approximately half the patients in the study had reached 24 weeks of treatment.
Both OCREVUS subcutaneous injection and OCREVUS IV infusion resulted in rapid and near-complete suppression of MRI lesion activity by 24 weeks, with most patients having no T1 gadolinium-enhancing (T1-Gd+) lesions, which are markers of active inflammation, and no new/enlarging T2 lesions, which represent the amount of disease burden or lesion load at 24 weeks.
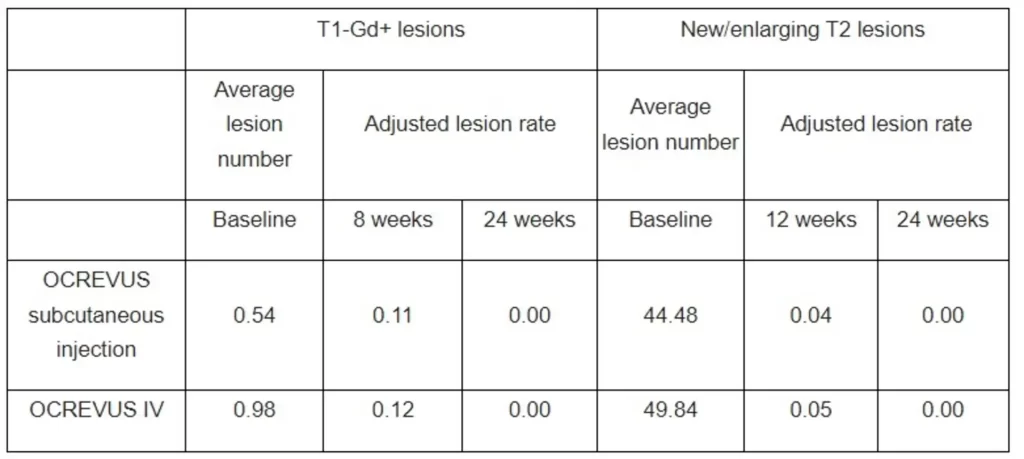
The safety profile of OCREVUS subcutaneous injection was consistent with the well-established safety profile of OCREVUS IV infusion. No new safety signals were identified for OCREVUS subcutaneous injection. The most common adverse events in the OCREVUS subcutaneous injection group were injection reactions (48% of all exposed patients), all of which were either mild or moderate. The most common AEs in the OCREVUS IV infusion group were infusion-related reactions (17%). A total of 4 and 7 serious AEs were experienced by 3 (2.5%) and 4 (3.4%) patients in the OCREVUS subcutaneous and IV infusion groups, respectively.
The OCREVUS twice-yearly, 10-minute subcutaneous injection is healthcare provider administered and designed to be delivered without the need for IV infrastructure, so it has the potential to expand the usage of OCREVUS in treatment centres without IV infrastructure or those with IV capacity limitations. This provides an additional delivery option so that OCREVUS can be matched to the individual needs of people with MS and healthcare professionals.
The OCARINA II data will be submitted to health authorities around the world in the coming months. Roche is committed to advancing innovative clinical research programmes to broaden the scientific understanding of MS, further reduce disability progression in RMS and PPMS and improve the treatment experiences for those living with the disease.
The investigational subcutaneous formulation combines OCREVUS with Halozyme Therapeutics’ Enhanze® drug delivery technology.
OCREVUS is a humanised monoclonal antibody designed to target CD20-positive B cells, a specific type of immune cell thought to be a key contributor to myelin (nerve cell insulation and support) and axonal (nerve cell) damage. This nerve cell damage can lead to disability in people with MS. Based on preclinical studies, OCREVUS binds to CD20 cell surface proteins expressed on certain B cells, but not on stem cells or plasma cells, suggesting that important functions of the immune system may be preserved.
The Enhanze drug delivery technology is based on a proprietary recombinant human hyaluronidase PH20 (rHuPH20), an enzyme that locally and temporarily degrades hyaluronan – a glycosaminoglycan or chain of natural sugars in the body – in the subcutaneous space. This increases the permeability of the tissue under the skin, allowing space for large molecules like OCREVUS to enter, and enables the subcutaneous formulation to be rapidly dispersed and absorbed into the bloodstream.
OCREVUS IV is the first and only therapy approved for both RMS (including relapsing-remitting MS [RRMS] and active, or relapsing secondary progressive MS [SPMS], in addition to clinically isolated syndrome [CIS] in the U.S.) and PPMS. OCREVUS IV is administered by intravenous infusion every six months. The initial dose is given as two 300 mg infusions given two weeks apart. Subsequent doses are given as single 600 mg infusions.
OCARINA II is a Phase III, global, multicentre, randomised study evaluating the pharmacokinetics, safety and radiological and clinical effects of the subcutaneous formulation of OCREVUS compared with OCREVUS intravenous (IV) infusion in 236 patients with relapsing MS (RMS) or primary progressive MS (PPMS). The primary endpoint is non-inferiority in area under the serum concentration time curve (AUC) from day 1 to 12 weeks after subcutaneous injection compared to IV infusion. Secondary endpoints include maximum serum concentration (Cmax) of OCREVUS, the total number of active, gadolinium-enhancing T1 lesions at 8 and 12 weeks, and new or enlarging T2 lesions at 12 and 24 weeks, as well as safety and immunogenicity outcomes. Exploratory endpoints include patient-reported outcomes.
Multiple sclerosis (MS) is a chronic disease that affects more than 2.8 million people worldwide. MS occurs when the immune system abnormally attacks the insulation and support around nerve cells (myelin sheath) in the central nervous system (brain, spinal cord and optic nerves), causing inflammation and consequent damage. This damage can cause a wide range of symptoms, including muscle weakness, fatigue and difficulty seeing, and may eventually lead to disability. Most people with MS experience their first symptom between 20 and 40 years of age, making the disease the leading cause of non-traumatic disability in younger adults.
People with all forms of MS experience disease progression – permanent loss of nerve cells in the central nervous system – from the beginning of their disease even if their clinical symptoms aren’t apparent or don’t appear to be getting worse. Delays in diagnosis and treatment can negatively impact people with MS, in terms of their physical and mental health, and contribute to the negative financial impact on the individual and society. An important goal of treating MS is to slow, stop and ideally prevent disease activity and progression as early as possible.
Relapsing-remitting MS (RRMS) is the most common form of the disease and is characterised by episodes of new or worsening signs or symptoms (relapses) followed by periods of recovery. Approximately 85% of people with MS are initially diagnosed with RRMS. The majority of people who are diagnosed with RRMS will eventually transition to secondary progressive MS (SPMS), in which they experience steadily worsening disability over time. Relapsing forms of MS (RMS) include people with RRMS and people with SPMS who continue to experience relapses. Primary progressive MS (PPMS) is a debilitating form of the disease marked by steadily worsening symptoms but typically without distinct relapses or periods of remission. Approximately 15% of people with MS are diagnosed with the primary progressive form of the disease. Until the FDA approval of OCREVUS, there had been no FDA-approved treatments for PPMS.
Neuroscience is a major focus of research and development at Roche. Our goal is to pursue ground-breaking science to develop new treatments that help improve the lives of people with chronic and potentially devastating diseases. Roche and Genentech are investigating more than a dozen medicines for neurological disorders, including MS, spinal muscular atrophy, neuromyelitis optica spectrum disorder, Alzheimer’s disease, Huntington’s disease, Parkinson’s disease, acute ischemic stroke, Duchenne muscular dystrophy and Angelman syndrome. Together with our partners, we are committed to pushing the boundaries of scientific understanding to solve some of the most difficult challenges in neuroscience today.
Roche is a global pioneer in pharmaceuticals and diagnostics focused on advancing science to improve people’s lives. The combined strengths of pharmaceuticals and diagnostics under one roof have made Roche the leader in personalised healthcare – a strategy that aims to fit the right treatment to each patient in the best way possible.
Roche is the world’s largest biotech company, with truly differentiated medicines in oncology, immunology, infectious diseases, ophthalmology and diseases of the central nervous system. Roche is also the world leader in in vitro diagnostics and tissue-based cancer diagnostics, and a frontrunner in diabetes management.
Founded in 1896, Roche continues to search for better ways to prevent, diagnose and treat diseases and make a sustainable contribution to society. The company also aims to improve patient access to medical innovations by working with all relevant stakeholders. More than thirty medicines developed by Roche are included in the World Health Organization Model Lists of Essential Medicines, among them life-saving antibiotics, antimalarials and cancer medicines. Moreover, for the twelfth consecutive year, Roche has been recognised as one of the most sustainable companies in the Pharmaceuticals Industry by the Dow Jones Sustainability Indices (DJSI).
The Roche Group, headquartered in Basel, Switzerland, is active in over 100 countries and in 2020 employed more than 100,000 people worldwide. In 2020, Roche invested CHF 12.2 billion in R&D and posted sales of CHF 58.3 billion. Genentech, in the United States, is a wholly owned member of the Roche Group. Roche is the majority shareholder in Chugai Pharmaceutical, Japan
New data from Phase II FENopta study in relapsing multiple sclerosis (RMS) show fenebrutinib crosses the blood-brain barrier with the potential to act directly on the chronic inflammation related to multiple sclerosis (MS)
More than 90% relative reduction in new/enlarging T2 lesions and new T1 gadolinium-enhancing (Gd+) lesions with fenebrutinib beginning at 8 weeks
The safety profile of fenebrutinib was consistent with previous and ongoing clinical trials across more than 2,500 people to date
Roche (SIX: RO, ROG; OTCQX: RHHBY) today announced new data from the Phase II FENopta study showing that investigational, oral fenebrutinib is brain penetrant and reduces brain lesions in people with relapsing multiple sclerosis (RMS) with a consistent safety profile to other fenebrutinib trials. The late-breaking data were featured in an oral presentation at the 9th Joint ECTRIMS-ACTRIMS Meeting (European and Americas Committees for Treatment and Research in Multiple Sclerosis).
“These interesting results raise the possibility that fenebrutinib slows MS disease progression in part by acting directly within the brain,” said Levi Garraway, M.D., Ph.D., Roche’s Chief Medical Officer and Head of Global Product Development. “These data, which we are currently confirming in pivotal trials of both relapsing and progressive MS, suggest that fenebrutinib may have the potential to counteract acute and chronic inflammation within the brain to reduce disease activity in people with MS.”
Brain penetrance was measured by the level of fenebrutinib in the cerebrospinal fluid (CSF) of a subgroup of 11 patients with RMS. After 12 weeks of continuous treatment, the mean fenebrutinib concentration was 43.1 ng/mL. Similar fenebrutinib concentrations can produce near-maximal inhibition (IC90) in preclinical studies. Thus, the level of fenebrutinib in the brain and central nervous system may conceivably become high enough to reduce MS disease activity and progression in patients.
Fenebrutinib significantly reduced the total number of new T1 gadolinium-enhancing (T1 Gd+) brain lesions which are markers of active inflammation, and the total number of new or enlarging T2-weighted (T2) brain lesions, which represent the amount of disease burden or chronic lesion load. A rapid onset of lesion reduction was observed by 4 weeks, with relative reductions of 92% and 90% in T1 Gd+ lesions and relative reductions of 90% and 95% in T2 lesions observed at 8 and 12 weeks, respectively.
Furthermore, patients treated with fenebrutinib were four times more likely to be free from any new T1 Gd+ brain lesions and new or enlarging T2 brain lesions at weeks 4, 8, and 12 combined, compared to patients who received placebo (odds ratio 4.005, p=0.0117).
The safety profile of fenebrutinib was consistent with previous and ongoing fenebrutinib clinical trials across more than 2,500 people to date. There were no new safety concerns identified in the FENopta study. Overall rates of adverse events were 38% for fenebrutinib and 33% for placebo. The most common adverse events that were higher with fenebrutinib than placebo were abnormal liver enzyme levels (5.5% fenebrutinib, 0% placebo), headache (4.1% fenebrutinib, 2.8% placebo), nasopharyngitis (2.7% fenebrutinib, 0% placebo) and upper abdominal pain (2.7% fenebrutinib, 0% placebo).
Fenebrutinib is the only non-covalent and reversible BTK inhibitor in Phase III trials for MS and was designed to be highly selective, which may be important in reducing off-target effects of a molecule and potentially contribute to long-term safety outcomes. An open-label extension of FENopta is ongoing, with Phase III studies FENhance 1 and 2 currently enrolling patients with RMS and FENtrepid fully enrolled for patients with primary progressive MS (PPMS). Roche is committed to advancing innovative clinical research programmes to broaden the scientific understanding of MS, further reduce disability worsening in RMS and PPMS and improve the treatment experiences for those living with the disease.
Fenebrutinib is an investigational oral, reversible and non-covalent Bruton’s tyrosine kinase (BTK) inhibitor that blocks the function of BTK. BTK, also known as tyrosine-protein kinase BTK, is an enzyme that regulates B-cell development and activation and is also involved in the activation of innate immune system myeloid lineage cells, such as macrophages and microglia. Preclinical data have shown fenebrutinib to be potent and highly selective, and it is the only reversible inhibitor currently in Phase III trials for MS. Fenebrutinib has been shown to be 130 times more selective for BTK vs. other kinases. These design features may be important as the high selectivity and reversibility can potentially reduce off-target effects of a molecule.
Fenebrutinib is a dual inhibitor of both B-cell and microglia activation. This dual inhibition may be able to reduce both MS disease activity and disability progression, thereby potentially addressing the key unmet medical need in people living with MS. The Phase III programme includes two identical trials in RMS (FENhance 1 & 2) with an active teriflunomide comparator and one trial in PPMS (FENtrepid) in which fenebrutinib is being evaluated against OCREVUS® (ocrelizumab). To date, more than 2,500 patients and healthy volunteers have been treated with fenebrutinib in Phase I, II and III clinical programmes across multiple diseases, including MS and other autoimmune disorders.
The FENopta study is a global Phase II, randomised, double-blind, placebo-controlled 12-week study to investigate the efficacy, safety and pharmacokinetics of fenebrutinib in 109 adults aged 18-55 years with RMS. The primary endpoint is the total number of new gadolinium-enhancing T1 lesions as measured by MRI scans of the brain at 4, 8 and 12 weeks. Secondary endpoints include the number of new or enlarging T2-weighted lesions as measured by MRI scans of the brain at 4, 8 and 12 weeks, and the proportion of patients free from any new gadolinium-enhancing T1 lesions and new or enlarging T2-weighted lesions as measured by MRI scans of the brain at 4, 8 and 12 weeks. The goal of the FENopta study is to characterise the effect of fenebrutinib on MRI and soluble biomarkers of disease activity and progression, and it includes an optional substudy to measure cerebrospinal fluid biomarkers of neuronal injury. Patients who complete the double-blind period are eligible for an open-label extension study.
Multiple sclerosis (MS) is a chronic disease that affects more than 2.8 million people worldwide. MS occurs when the immune system abnormally attacks the insulation and support around nerve cells (myelin sheath) in the central nervous system (brain, spinal cord and optic nerves), causing inflammation and consequent damage. This damage can cause a wide range of symptoms, including muscle weakness, fatigue and difficulty seeing, and may eventually lead to disability. Most people with MS experience their first symptom between 20 and 40 years of age, making the disease the leading cause of non-traumatic disability in younger adults.
People with all forms of MS experience disease progression – permanent loss of nerve cells in the central nervous system – from the beginning of their disease even if their clinical symptoms aren’t apparent or don’t appear to be getting worse. Delays in diagnosis and treatment can negatively impact people with MS, in terms of their physical and mental health, and contribute to the negative financial impact on the individual and society. An important goal of treating MS is to slow, stop and ideally prevent disease activity and progression as early as possible.
Relapsing-remitting MS (RRMS) is the most common form of the disease and is characterised by episodes of new or worsening signs or symptoms (relapses) followed by periods of recovery. Approximately 85% of people with MS are initially diagnosed with RRMS. The majority of people who are diagnosed with RRMS will eventually transition to secondary progressive MS (SPMS), in which they experience steadily worsening disability over time. Relapsing forms of MS (RMS) include people with RRMS and people with SPMS who continue to experience relapses. Primary progressive MS (PPMS) is a debilitating form of the disease marked by steadily worsening symptoms but typically without distinct relapses or periods of remission. Approximately 15% of people with MS are diagnosed with the primary progressive form of the disease. Until the FDA approval of OCREVUS, there had been no FDA-approved treatments for PPMS.
Neuroscience is a major focus of research and development at Roche. Our goal is to pursue ground-breaking science to develop new treatments that help improve the lives of people with chronic and potentially devastating diseases. Roche and Genentech are investigating more than a dozen medicines for neurological disorders, including MS, spinal muscular atrophy, neuromyelitis optica spectrum disorder, Alzheimer’s disease, Huntington’s disease, Parkinson’s disease, acute ischemic stroke, Duchenne muscular dystrophy and Angelman syndrome. Together with our partners, we are committed to pushing the boundaries of scientific understanding to solve some of the most difficult challenges in neuroscience today.
Roche is a global pioneer in pharmaceuticals and diagnostics focused on advancing science to improve people’s lives. The combined strengths of pharmaceuticals and diagnostics under one roof have made Roche the leader in personalised healthcare – a strategy that aims to fit the right treatment to each patient in the best way possible.
Roche is the world’s largest biotech company, with truly differentiated medicines in oncology, immunology, infectious diseases, ophthalmology and diseases of the central nervous system. Roche is also the world leader in in vitro diagnostics and tissue-based cancer diagnostics, and a frontrunner in diabetes management.
Founded in 1896, Roche continues to search for better ways to prevent, diagnose and treat diseases and make a sustainable contribution to society. The company also aims to improve patient access to medical innovations by working with all relevant stakeholders. More than thirty medicines developed by Roche are included in the World Health Organization Model Lists of Essential Medicines, among them life-saving antibiotics, antimalarials and cancer medicines. Moreover, for the twelfth consecutive year, Roche has been recognised as one of the most sustainable companies in the Pharmaceuticals Industry by the Dow Jones Sustainability Indices (DJSI).
The Roche Group, headquartered in Basel, Switzerland, is active in over 100 countries and in 2020 employed more than 100,000 people worldwide. In 2020, Roche invested CHF 12.2 billion in R&D and posted sales of CHF 58.3 billion. Genentech, in the United States, is a wholly owned member of the Roche Group. Roche is the majority shareholder in Chugai Pharmaceutical, Japan
Vabysmo showed robust and sustained retinal drying up to 72 weeks and a safety profile consistent with previous studies
Regulatory applications for Vabysmo in RVO are under review by health authorities around the world; if approved, RVO would be the third indication in addition to nAMD and DME
Vabysmo is the first and only treatment that targets and inhibits two signalling pathways linked to a number of vision-threatening retinal conditions
Basel, 10 October 2023 - Roche (SIX: RO, ROG; OTCQX: RHHBY) today announced positive topline long-term results from the global phase III BALATON and COMINO studies, evaluating extended treatment intervals with Vabysmo® (faricimab) in macular edema due to branch and central retinal vein occlusion (BRVO and CRVO).1,2
From weeks 24 to 72, all people in both studies received Vabysmo using a treat-and-extend dosing regimen, which allows tailoring of their treatment interval according to the individual patient's response to treatment. Data showed that people treated with Vabysmo extended their treatment intervals up to every four months while maintaining the vision gains achieved in the first 24 weeks of the studies. Vabysmo continued to show robust and sustained drying of retinal fluid from baseline up to week 72, as measured by reduction in central subfield thickness. This is the first time that vision and anatomical improvements have been maintained for more than a year using a personalised treat-and-extend dosing regimen in global phase III studies for both BRVO and CRVO. In both studies, Vabysmo was generally well-tolerated and the safety profile was consistent with previous studies.
“These are the first retinal vein occlusion (RVO) studies to show vision maintenance and anatomical improvements up to 72 weeks in both central and branch RVO,” said Levi Garraway, M.D., Ph.D., Roche’s Chief Medical Officer and Head of Global Product Development. “These data further support Vabysmo’s potential as a new treatment for RVO, allowing people to preserve their vision while spending less time managing their condition.”
RVO impacts 28 million people globally and, if approved, would be the third indication for Vabysmo in addition to neovascular or ‘wet’ age-related macular degeneration (nAMD) and diabetic macular edema (DME).3-7 Together, the three conditions affect around 70 million people worldwide and are among the leading causes of vision loss.
Detailed results from weeks 24 to 72 of the phase III BALATON and COMINO studies will be presented at an upcoming medical meeting.
Data from the first 24 weeks of the phase III BALATON and COMINO studies, presented at Angiogenesis, Exudation and Degeneration 2023, demonstrated early and sustained vision improvement with Vabysmo, with both studies meeting their primary endpoints of non-inferior vision gains compared to aflibercept. A secondary endpoint showed that Vabysmo achieved rapid and robust drying of retinal fluid from baseline to week 24, as measured by reduction in central subfield thickness.12
Data up to 24 weeks have been submitted to global health authorities, including the United States Food and Drug Administration (U.S. FDA) and European Medicines Agency. A decision from the U.S. FDA is expected in late 2023. Vabysmo is uniquely engineered to target and inhibit two signalling pathways, which are linked to a number of vision-threatening retinal conditions, by neutralising angiopoietin-2 (Ang-2) and vascular endothelial growth factor-A (VEGF-A) to restore vascular stability.13,14 The level of Ang-2 is elevated in RVO and it is thought that increased Ang-2 expression drives disease progression.
To date, Vabysmo is approved in more than 80 countries around the world for people living with nAMD and DME, including the United States, Japan, the United Kingdom and the European Union, with public reimbursement in over 25 markets and more than 1.5 million doses distributed globally.
RVO is the second most common cause of vision loss due to retinal vascular diseases. It affects an estimated 28 million adults globally, mainly those aged 60 or older, and can lead to severe and sudden vision loss.3,17 The level of angiopoietin-2 (Ang-2) is elevated in RVO and it is thought that increased Ang-2 expression drives disease progression.11,15 RVO typically results in sudden, painless vision loss in the affected eye because the vein blockage restricts normal blood flow in the affected retina, resulting in ischaemia, bleeding, fluid leakage and retinal swelling called macular edema.17-19 Currently, macular edema due to RVO is typically treated with repeated intravitreal injections of anti-vascular endothelial growth factor therapies.18 There are two main types of RVO: branch retinal vein occlusion (BRVO), which affects more than 23 million people globally and occurs when one of the four smaller ‘branches’ of the main central retinal vein becomes blocked; and central retinal vein occlusion (CRVO), which is less common, affecting more than four million people worldwide, and occurs when the eye’s central retinal vein becomes blocked.
BALATON (NCT04740905) and COMINO (NCT04740931) are two randomised, multicentre, double-masked, global phase III studies evaluating the efficacy and safety of Vabysmo®️ (faricimab) compared to aflibercept. For the first 20 weeks, patients were randomised 1:1 to receive six-monthly injections of either Vabysmo (6.0 mg) or aflibercept (2.0 mg). From weeks 24 to 72, all patients received Vabysmo (6.0 mg) up to every four months, using a treat-and-extend dosing regimen.
The BALATON study was conducted in 553 people with branch retinal vein occlusion. The COMINO study was conducted in 729 people with central retinal or hemiretinal vein occlusion. The primary endpoint of each study was the change in best-corrected visual acuity from baseline at 24 weeks. Secondary endpoints (weeks 0-24) included change in central subfield thickness and drying of retinal fluid, from baseline over time up to week 24. Secondary endpoints (weeks 24-72) were treatment durability at 68 weeks and continuation of weeks 0-24 endpoints.
Roche has a robust phase III clinical development programme for Vabysmo. The programme includes AVONELLE-X, an extension study of TENAYA and LUCERNE, evaluating the long-term safety and tolerability of Vabysmo in neovascular or ‘wet’ age-related macular degeneration (nAMD), and Rhone-X, an extension study of YOSEMITE and RHINE, evaluating the long-term safety and tolerability of Vabysmo in diabetic macular edema (DME).20.21 Roche has also initiated several phase IV studies, including the ELEVATUM study of Vabysmo in underrepresented patient populations with DME, the SALWEEN study of Vabysmo in a subpopulation of nAMD highly prevalent in Asia, as well as the VOYAGER study, a global real-world data collection platform.22-24 Roche also supports several other independent studies to further understand retinal conditions with a high unmet need.
Vabysmo is the first bispecific antibody approved for the eye.4,6 It targets and inhibits two signalling pathways linked to a number of vision-threatening retinal conditions by neutralising angiopoietin-2 (Ang-2) and vascular endothelial growth factor-A (VEGF-A). Ang-2 and VEGF-A contribute to vision loss by destabilising blood vessels, causing new leaky blood vessels to form and increasing inflammation. By blocking pathways involving Ang-2 and VEGF-A, Vabysmo is designed to stabilise blood vessels.13,14 Vabysmo is approved in more than 80 countries around the world, including the United States, Japan, the United Kingdom and the European Union for people living with neovascular or ‘wet’ age-related macular degeneration and diabetic macular edema. Review by other regulatory authorities is ongoing.
Roche is focused on saving people’s eyesight from the leading causes of vision loss through pioneering therapies. Through our innovation in the scientific discovery of new potential drug targets, personalised healthcare, molecular engineering, biomarkers and continuous drug delivery, we strive to design the right therapies for the right patients.
We have the broadest retina pipeline in ophthalmology, which is led by science and informed by insights from people with eye diseases. Our pipeline includes gene therapies and treatments for geographic atrophy and other vision-threatening diseases, including rare and inherited conditions.
Applying our extensive experience, we have already brought breakthrough ophthalmic treatments to people living with vision loss. Susvimo™ (previously called Port Delivery System with ranibizumab) 100 mg/mL for intravitreal use via ocular implant is the first United States Food and Drug Administration-approved refillable eye implant for neovascular or ‘wet’ age-related macular degeneration that continuously delivers a customised formulation of ranibizumab over a period of months.25 Vabysmo® (faricimab) is the first bispecific antibody approved for the eye, which targets and inhibits two signalling pathways linked to a number of vision-threatening retinal conditions.4,6,13,14 Lucentis® (ranibizumab injection)^ is the first treatment approved to improve vision in people with certain retinal conditions.
Roche is a global pioneer in pharmaceuticals and diagnostics focused on advancing science to improve people’s lives. The combined strengths of pharmaceuticals and diagnostics under one roof have made Roche the leader in personalised healthcare – a strategy that aims to fit the right treatment to each patient in the best way possible.
Roche is the world’s largest biotech company, with truly differentiated medicines in oncology, immunology, infectious diseases, ophthalmology and diseases of the central nervous system. Roche is also the world leader in in vitro diagnostics and tissue-based cancer diagnostics, and a frontrunner in diabetes management.
Founded in 1896, Roche continues to search for better ways to prevent, diagnose and treat diseases and make a sustainable contribution to society. The company also aims to improve patient access to medical innovations by working with all relevant stakeholders. More than thirty medicines developed by Roche are included in the World Health Organization Model Lists of Essential Medicines, among them life-saving antibiotics, antimalarials and cancer medicines. Moreover, for the twelfth consecutive year, Roche has been recognised as one of the most sustainable companies in the Pharmaceuticals Industry by the Dow Jones Sustainability Indices (DJSI).
The Roche Group, headquartered in Basel, Switzerland, is active in over 100 countries and in 2020 employed more than 100,000 people worldwide. In 2020, Roche invested CHF 12.2 billion in R&D and posted sales of CHF 58.3 billion. Genentech, in the United States, is a wholly owned member of the Roche Group. Roche is the majority shareholder in Chugai Pharmaceutical, Japan